

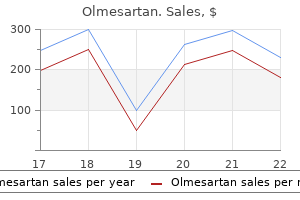
Buy generic olmesartan 10 mg online
However hypertension kidney specialists lancaster pa buy generic olmesartan online, sperm banking in puberty or prepuberty presents ethical concerns as to the procedure, the cost of banking, and the uncertainty of outcome and is not presently standard therapy. Associated cortisol deficiency and increased mineralocorticoid secretion in this condition lead to hypertension, decreased serum potassium levels, and metabolic alkalosis. Elevated serum progesterone levels and decreased plasma renin activity are helpful diagnostic features. Death often occurs in infancy if untreated because of unrecognized glucocorticoid and mineralocorticoid deficiencies. Cryptorchidism is the condition in which one or both testes have not reached the bottom of the scrotum before birth. When testes are not descended they may be located in high scrotal, suprascrotal, or inguinal positions or can be nonpalpable, which includes ectoptic testes as well. Testes may ascend after birth, ascensus testis, which leads to a higher prevalence of undescended testes in prepuberty than at birth in several international reports, and these patients are not always included in cryptorchidism surveys. Study of more than 1 million Danish boys showed the concordance rate of cryptorchidism was 3. About 50% of bilateral, nonpalpable testes are undescended, and the other 50% are testicular remnants from vanishing testes that usually do not contain germ cells, are found in the scrotum, are not at risk for carcinoma, and need not be removed if the history is certain. Serum gonadotropins follow the normal Ushaped curve of high values in infancy and puberty with lower values in midchildhood in anorchia, although the values are above normal. Compensatory hypertrophy occurs if there is no contralateral testis, and this aids the diagnosis. A Japanese study demonstrated that the mean contralateral testicular length and volume in the boys with an absent testis were 22. If there is an unilateral undescended testis, the increased relative risk of carcinoma in the contralateral descended testis is 1. There is no compelling evidence that hor- monal therapy in the short term is harmful, but unsuccessful medical therapy should not be allowed to significantly delay surgical therapy. The test was positive in Prader-Willi syndrome when the maximum testosterone level after 72 hours was 2 to 20 times higher than baseline levels in infants aged 3 to 12 months and 5 to 10 times higher, between 2. Because testicular descent normally occurs by 1 year of age, orchidopexy is recommended between 6 and 12 months in those whose have testes that do not descend with medical treatment or upon discovering cryptorchidism after 12 months of age. Cryptorchid testes may demonstrate congenital abnormalities and may not function normally even if brought into the scrotum early in life. Two critical steps in the maturation of germ cells are described in the normal prepubertal testis that do not occur in the unilaterally undescended testes. The contralateral descended testis is affected but less so than the undescended testis. Identification of the gonocyte transformation has influenced recommendations on the timing of orchidopexy. Postpubertal orchidopexy is associated with a greater than 85% prevalence of azoospermia or oligospermia. It has been surmised that cryptorchid testes, even if replaced in the scrotum, may never have normal spermatogenic function as a consequence of an early abnormality in germ cell maturation, vascular damage to the testicular circulation during orchidopexy, or an intrinsic testicular defect. Fertility potential varies depending upon preoperative history and laboratory results. Testicular dysgenesis may be indicated by increased gonadotropin levels and early surgery may be indicated; normal gonadotropin levels and a decreased germ cell number, may indicate transient hypothalamus-pituitarygonadal hypofunction with a poor fertility prognosis; and if there are normal gonadotropins, inhibin B, and germ cell number, there is a good fertility prognosis. Cryptorchidism is associated with an increased risk of cancer of the testes, which is rising. The incidence of testicular carcinoma in England at all ages increased from 2 per 100,000 in 1909 to 4. Undescended testes remain at a higher temperature than descended testes, and undescended testes have a maturation arrest at the conversion of the gonocytes to spermatogonia, which appears to direct the testes toward malignant degeneration. There is a very small risk of carcinoma of the testes in prepuberty, but the absence of carcinoma in situ in prepuberty is not an assurance that carcinoma will not develop in adult life. Periodic sonography of the testis of affected patients is recommended after the onset of puberty. They are considered a normal variation, but a requirement for orchiopexy was reported for 22. The finding of one case of testicular carcinoma in a boy with spontaneous descent in this series led to a suggestion of following such cases in the long term. The 45,X karyotype is associated with female phenotype, short stature, sexual infantilism, and various somatic abnormalities. Sex chromosome mosaicism or structural abnormalities of an X or Y chromosome (affects about 40% of individuals with Turner syndrome) may modify the features of this syndrome. The syndrome of gonadal dysgenesis and its variants are found in a continuum ranging from the typical 45,X phenotype to a normal male or female phenotype. The 45,X abortuses have edema and large hygromas of the neck that may be seen on prenatal ultrasound studies. This lymphatic defect is the basis for the loose skinfolds that ultimately form the webbed neck. Affected newborn infants may also have lymphedema of the extremities; the term Bonnevie-Ullrich syndrome has been applied to newborn infants with these features of Turner syndrome. Frequent features are distinct facies with micrognathia, a fish-mouth appearance, high-arched palate with dental abnormalities, epicanthal folds, ptosis, low-set or deformed ears, short neck with low hairline, webbing. A broad, shieldlike chest leads to the appearance of widely spaced nipples, and the areolae are often hypoplastic. Skeletal defects include short fourth metacarpals and cubitus valgus (which may develop after birth), Madelung deformity of the wrist (in about 7%), genu valgum, and scoliosis. There are extensive pigmented nevi,562 a tendency to keloid formation, and hypoplastic nails. Lymphatic obstruction leads to the infantile puffiness of extremities and pterygium colli and to a distinctive shape of the ears. Cardiovascular anomalies affect the left side of the heart and include coarctation of the aorta in about 10% (40% of these have associated webbing of the neck), aortic stenosis, and bicuspid aortic valves; the latter individuals are at risk for a dissecting aortic aneurysm. She exhibited characteristic stigmata of the syndrome: a short, webbed neck; shield-like chest with widely separated nipples; bilateral metacarpal signs; puffiness over the dorsum of the fingers; cubitus valgus; increased number of pigmented nevi; characteristic facies; and low-set ears. Vaginal smears and the urocytogram showed an immature pattern in which cornified squamous cells were absent. Female secondary sexual characteristics were induced with estrogen therapy, and the cyclic administration resulted in periodic estrogen withdrawal bleeding. Apart from short stature (height, 118 cm; height age, 6 years and 11 months), increased pigmented nevi, and subtle changes in the fingers and toes, she had few somatic anomalies. In contrast to the patient on the left, the main clinical feature was short stature. Defects of the gastrointestinal system include intestinal telangiectasias and hemangiomatoses that rarely can lead to massive gastrointestinal bleeding. The prevalence of inflammatory bowel disease, chronic liver disease, and colon cancer is increased. Autoimmune diseases, such as Hashimoto thyroiditis (16-fold relative risk) and Graves disease, are common, and an association with juvenile rheumatoid arthritis and psoriatic arthritis has been described. The age of diagnosis of Turner syndrome continues to be delayed, with the exception of newborns with the striking phenotype of the Bonnevie-Ullrich syndrome or those diagnosed on amniocentesis. Ultrasensitive estrogen bioassays can confirm decreased ovarian function in girls with Turner syndrome because estradiol values are significantly lower than those found in average girls in puberty. Affected adults can undergo hormone replacement to prepare the uterus to receive a donated embryo and proceed to delivery. Decreased growth rate occurs at the time of expected puberty, and the pubertal growth spurt is absent in those without pubertal development. Untreated individuals with Turner syndrome in the United Kingdom and United States have a mean adult height of approximately 142 to 143 cm, which is about 20 cm less than the average height of typical women; the adult stature of these patients correlates with midparental height and with the height of unaffected women of the same ethnic group. It is postulated that a second gene on the short arm of the X chromosome that does not undergo X-chromosome inactivation contributes the other one third of the deficit. In girls with Turner syndrome with spontaneous puberty, pubertal height velocity was transiently higher than in girls with amenorrhea, but adult height was not different.
Purchase 10 mg olmesartan with amex
For microvascular disease end points hypertension 130100 order 40 mg olmesartan, there is an almost 10-fold increase in risk as HbA1c increases from 5. Evidence of such selective vascular resistance to insulin has been demonstrated in the obese Zucker rat. Some subpopulations of macrophages are proinflammatory, while others are anti-inflammatory. Macrophages isolated from two different mouse models of type 1 diabetes exhibit a proinflammatory phenotype. Increased flux of fatty acids from insulin-resistant adipose tissue to arterial cells both indirectly via endothelial catabolism of triglyceride-rich lipoproteins98 or directly may be such a consequence. Normally, in response to acute ischemia, new blood vessel growth rescues stunned areas of the heart or central nervous system, reducing morbidity and mortality risks. In response to chronic ischemia, collateral vessel development reduces the size and severity of subsequent infarction. In response to ischemia, circulating endothelial progenitor cells from the bone marrow promote the regeneration of blood vessels, acting in concert with cells and extracellular matrix at the site of injury. In experimental diabetes, however, these circulating endothelial progenitor cells are depleted and dysfunctional. As a result, diabetic animals have decreased vascular density after hind limb ischemia. Similarly, in human diabetes, endothelial progenitor cells are also depleted and dysfunctional. This results in part from a failure to form adequate compensatory microvascu- lature in response to ischemia. Posttranslational modification of proteins by the glycolysis-derived -oxoaldehyde, methylglyoxal, appears to play a central role in this failure. They respond normally to ischemia, in contrast to ischemic tissue elsewhere in the body. Mechanisms of Hyperglycemia-Induced Damage Murine models are valuable tools for defining the pathogenesis of diabetic complications. However, they have significant limitations, and it is important to recognize some of their limits. No diabetic animal model, regardless of genetic background, recapitulates the structural and functional alterations of human complications. Consistent with this is the finding from a cross-species comparison of glomerular transcriptional networks from patients with diabetic nephropathy with those from three diabetic mouse models. This study showed that gene expression changes in these mouse models are similar to those in human nephropathy before the development of microalbuminuria, and therefore are most relevant to changes in very early human diabetic nephropathy. Most of the mechanistic data currently available come from studies of the early stages of each complication. More data about what molecular mechanisms are dominant in each complication target tissue at each pathologic stage are urgently needed. In addition, only 33% to 50% of patients with poor glycemic control develop diabetic nephropathy, and a subset of patients with good glycemic control still develop diabetic nephropathy. Using primary human cells and mouse models, four major hypotheses about how hyperglycemia causes diabetic complications have generated a large amount of data as well as several clinical trials based on specific inhibitors of these mechanisms. From the late 1960s to 2000, there was no unifying hypothesis linking these four mechanisms together, nor was there an obvious connection between any of these mechanisms, each of which responds quickly to normalization of hyperglycemia, and the phenomenon of hyperglycemic memory (see earlier discussion). However, the amount of substrate converted to product per second (Kcat) of human aldose reductase for glucose is 0. Kcat values for most enzymes are between 1 and 104, although some have Kcat values orders of magnitude higher. Moreover, because the intracellular glucose concentration in capillary retinal endothelial cells incubated in 25 mM glucose is approximately 0. However, aldose reductase has high affinity and enzyme activity for a variety of other substrates, including several glycolytic intermediates such as glyceraldehyde 3-phosphate. Glyoxalase D-Lactate knockout mice, knockout of aldose reductase caused increased early lesion size in control and diabetic mice, rather than the expected decrease. Recent data show that in arteries of diabetic mice, aldose reductase drives hyperacetylation of Egr-1 with consequent upregulation of proinflammatory and prothrombotic signals. Glyoxal arises from the auto-oxidation of glucose, 3-deoxyglucosone arises from decomposition of the Amadori product, and methylglyoxal arises from fragmentation of glyceraldehyde 3-phosphate. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation end-product formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. In diabetic retinal capillaries, the earliest morphologic changes are pericyte loss and acellular capillary formation. The primary pathologic processes of retinal pericyte loss and acellular capillary formation are regulated by complex context-dependent interactions among a number of proangiogenic and antiangiogenic factors,132-134 including angiopoietin-2 (Ang-2). A similar mechanism involving methylglyoxal modification of other coregulator proteins may play a role in a variety of other diabetes-induced changes in gene expression. It also evokes thermal and mechanical hyperalgesia, which is reflected by increased blood flow in brain regions that are involved in pain processing. In nondiabetic mice, knockdown of Glo1 increases methylglyoxal modification of proteins and oxidative stress, causing alterations in kidney morphology indistinguishable from those caused by diabetes. In diabetic mice, Glo1 overexpression completely prevents diabetes-induced oxidative stress and kidney disease, despite unchanged levels of diabetic hyperglycemia. One codes for an isoform that lacks the amino-terminal V-type immunoglobulin-like domain (N-truncated), and one codes for an isoform lacking the carboxy-terminal transmembrane domain (C-truncated). Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. One of the most important emerging areas of hexosamine pathway investigation involves decreased contractility and altered calcium signaling. These issues were resolved by the discovery that each of the four different pathogenic mechanisms reflects a single hyperglycemia-induced process: overproduction of superoxide by the mitochondrial electron transport chain. When the electrochemical potential difference generated by this proton gradient is high, the life of superoxidegenerating electron transport intermediates such as ubisemiquinone is prolonged. Uncoupling proteins 2 and 3: potential regulators of mitochondrial energy metabolism. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. It has also been implicated in the pathogenesis of diabetic retinopathy and nephropathy in patients with type 2 diabetes. In the diabetic heart, increased fatty acid -oxidation can saturate the mitochondria, leading to myocardial steatosis, which may lead to cell dysfunction and death. In the retinas of diabetic animals with poor glycemic control for 6 months, subsequent normalization of HbA1c for 6 months had no effect on elevated retinal oxidative stress levels and only a small effect on elevated levels of 3-nitrotyrosine. Post-translational modifications of histones cause chromatin remodeling and changes in levels of gene expression. These epigenetic changes cause sustained increases in p65 gene expression and in the expression of p65-dependent proinflammatory genes. Both the epigenetic changes and the gene expression changes persist for at least 6 days of subsequent normal glycemia in cultured cells and for months in previously diabetic mice whose beta-cell function recovered. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that co-exist on the lysine tali. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. Monocytes from case subjects have statistically greater numbers of promoter regions with enrichment in H3K9Ac (active chromatin mark) compared with control subjects. Conventionally, 5mC is associated with a transcriptionally repressed chromatin state. Following streptozocin withdrawal, blood glucose and serum insulin return to physiologic levels as a result of pancreatic beta-cell regeneration. However, caudal fin regeneration and skin wound healing remain impaired, and this impairment is transmissible to daughter cell tissue. Despite decades of research, there is currently no known means of preventing diabetic retinopathy, and despite effective therapies, diabetic retinopathy remains the leading cause of new-onset blindness in working-age persons in most developed countries of the world. Diagnosis, management, and treatment of nonproliferative diabetic retinopathy and diabetic macular edema. These developments place further emphasis on the importance of adhering to lifelong routine ophthalmologic follow-up of the diabetic patient and optimization of associated systemic disorders. Rheologic changes occur in diabetic retinopathy and result from increased platelet aggregation, integrin-mediated leukocyte adhesion, and endothelial damage.
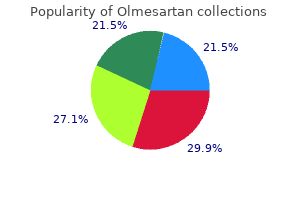
Buy cheap olmesartan 40 mg on-line
Metastases to the brain or skin may also be treated by palliative surgical resection arteria johnson buy 20mg olmesartan otc, but symptom relief is short lived and patients usually succumb to disease shortly after diagnosis. The surgeon must decide on the extent of dissection considering possible postoperative rehabilitation, quality of life, and life expectancy. The management decision involves discussions with other medical disciplines and especially the patient. Patients who progressed on the placebo could be crossed over to vandetanib at the time of tumor progression. Partial responses were observed in 45% of the patients randomized to vandetanib, and many patients were able to resume a normal quality of life. Only 12% of patients discontinued the drug due to toxicity, and 35% required dose reductions. There were significant side effects with the drug, including diarrhea, abdominal pain, fatigue, hypertension, and palmoplantar erythrodysesthesia, such that 16% of the patients discontinued the drug due to toxicity and 79% of patients required dose reductions. The cause of the diarrhea is unclear, but it can be debilitating in terms of quality of life and nutrition. Antimotility agents such as loperamide and codeine are often used as initial therapy. Patients with the syndrome are often debilitated due to hypokalemia, diabetes, hypertension, and gastritis. Ketoconazole, mifepristone, metyrapone, and mitotane have shown efficacy in some patients. Osteoporosis in multiple endocrine neoplasia type 1: severity, clinical significance, relationship to primary hyperparathyroidism, and response to parathyroidectomy. Heterogeneous size of the parathyroid glands in familial multiple endocrine neoplasia type 1. Primary and reoperative parathyroid operations in hyperparathyroidism of multiple endocrine neoplasia type 1. Low accuracy of tumor markers for diagnosing pancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1 patients. Parathyroid mitogenic activity in plasma from patients with familial multiple endocrine neoplasia type 1. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop. The superiority of minimally invasive parathyroidectomy based on 1650 consecutive patients with primary hyperparathyroidism. Reoperative parathyroid surgery in the era of sestamibi scanning and intraoperative parathyroid hormone monitoring. A prospective trial evaluating a standard approach to reoperation for missed parathyroid adenoma. Intraoperative parathormone measurement in patients with multiple endocrine neoplasia type I syndrome and hyperparathyroidism. Kinetic analysis of the rapid intraoperative parathyroid hormone assay in patients during operation for hyperparathyroidism. Sleeping parathyroid tumor: rapid hyperfunction after removal of the dominant tumor. The utility of routine transcervical thymectomy for multiple endocrine neoplasia 1-related hyperparathyroidism. Surgery for asymptomatic pancreatic lesion in multiple endocrine neoplasia type I. Prospective study of the clinical course, prognostic factors, causes of death, and survival in patients with long-standing Zollinger-Ellison syndrome. Pancreatic lesions and hormonal profile of pancreatic tumors in multiple endocrine neoplasia bleeding. The toxicity is most likely partly due to the dose of cabozantinib, as 60 mg/day has been the starting dose in subsequent clinical trials with other malignancies. Zur normalen und pathologischen histologie der glandula thyreoidea, parathyreoidea und hypophysis. Multiple endocrine adenomas; report of 8 cases in which the parathyroids, pituitary and pancreatic islets were involved. Concomitance of multiple adenomas of the parathyroids and pancreatic islets with tumor of the pituitary: a syndrome with a familial incidence. Multiplicity of hormone-secreting tumors: common themes about cause, expression, and management. The immediate upstream sequence of the mouse Ret gene controls tissue-specific expression in transgenic mice. Multiple endocrine neoplasia type I: assessment of laboratory tests to screen for the gene in a large kindred. Hyperparathyroidism presenting as the first lesion in multiple endocrine neoplasia type 1. Clinical and genetic investigation of a large kindred with multiple endocrine adenomatosis. Immunohistochemical study of 100 pancreatic tumors in 28 patients with multiple endocrine neoplasia, type I. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. Allelic deletions on chromosome 11q13 in multiple endocrine neoplasia type 1-associated and sporadic gastrinomas and pancreatic endocrine tumors. Precursor lesions in patients with multiple endocrine neoplasia type 1-associated duodenal gastrinomas. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Glucagon cell hyperplasia and neoplasia with and without glucagon receptor mutations. Multiple neuroendocrine tumors of the pancreas in von Hippel-Lindau disease patients: histopathological and molecular genetic analysis. Zollinger-Ellison syndrome can be the initial endocrine manifestation in patients with multiple endocrine neoplasia-type I. Clinical, anatomical, and evolutive features of patients with the Zollinger-Ellison syndrome combined with type I multiple endocrine neoplasia. Current concepts in the surgical management of multiple endocrine neoplasia type 1 pancreatic-duodenal disease. Results in the treatment of 40 patients with Zollinger-Ellison syndrome, hypoglycaemia or both. Pancreatectomy in multiple endocrine neoplasia type 1-related gastrinomas and pancreatic endocrine neoplasias. Outcome of duodenopancreatic resections in patients with multiple endocrine neoplasia type 1. Partial pancreaticoduodenectomy can provide cure for duodenal gastrinoma associated with multiple endocrine neoplasia type 1. Biochemically curative surgery for gastrinoma in multiple endocrine neoplasia type 1 patients. Gastric argyrophil carcinoidosis in patients with Zollinger-Ellison syndrome due to type 1 multiple endocrine neoplasia. The effect of Zollinger-Ellison syndrome and omeprazole therapy on gastric oxyntic endocrine cells. Prospective study of the antitumor efficacy of long-term octreotide treatment in patients with progressive metastatic gastrinoma. Familial hypocalciuric hypercalcemia: the relation to primary parathyroid hyperplasia. Noninvasive imaging of insulinomas and gastrinomas with endoscopic ultrasonography and somatostatin receptor scintigraphy. Localization of insulinomas to regions of the pancreas by intra-arterial stimulation with calcium. Development of resistance to a long-acting somatostatin analogue during treatment of two patients with metastatic endocrine pancreatic tumours. Increased parathyroid hormone-related peptide in patients with hypercalcemia associated with islet cell carcinoma. Acromegaly caused by growth hormone-relating hormone in a patient with multiple endocrine neoplasia type I.
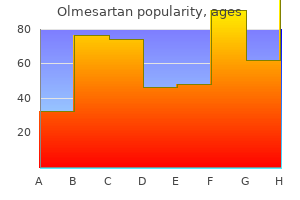
Order cheap olmesartan
Circulating growth factor levels are associated with tumorigenesis in neurofibromatosis type 1 untreated prehypertension cheap 10 mg olmesartan free shipping. Early puberty in internationally adopted girls: hormonal and clinical markers of puberty in 276 girls examined biannually over two years. A single histrelin implant is effective for 2 years for treatment of central precocious puberty. Use of a potent, long acting agonist of gonadotropin-releasing hormone in the treatment of precocious puberty. Suppression of the pituitarygonadal axis in children with central precocious puberty: effects on growth, growth hormone, insulin-like growth factor-I, and prolactin secretion. Long-term observation of 87 girls with idiopathic central precocious puberty treated with gonadotropin-releasing hormone analogs: impact on adult height, body mass index, bone mineral content, and reproductive function. Results of long-term follow-up after treatment of central precocious puberty with leuprorelin acetate: evaluation of effectiveness of treatment and recovery of gonadal function. Leydig-cell tumors caused by an activating mutation of the gene encoding the luteinizing hormone receptor [see comments]. Reduction of baseline body mass index under gonadotropin-suppressive therapy in girls with idiopathic precocious puberty. Random luteinizing hormone often remains pubertal in children treated with the histrelin implant for central precocious puberty. Boys with precocious puberty due to hypothalamic hamartoma: reproductive axis after discontinuation of gonadotropin-releasing hormone analog therapy. Prevalence of polycystic ovary syndrome in young women who had idiopathic central precocious puberty. Trait-specific tracking and determinants of body composition: a 7-year follow-up study of pubertal growth in girls. Slipped capital femoral epiphyses associated with the withdrawal of a gonadotrophin releasing hormone. Unexplained anaemia and failure to thrive as initial symptoms of infantile choriocarcinoma: a review. Precocious puberty associated with a pineal cyst: is it disinhibition of the hypothalamicpituitary axis Increased activation of the alternative "backdoor" pathway in patients with 21-hydroxylase deficiency: evidence from urinary steroid hormone analysis. Testicular adrenal rest tumors develop independently of long-term disease control: a longitudinal analysis of 50 adult men with congenital adrenal hyperplasia due to classic 21-hydroxylase deficiency. Leydig-cell tumour in children: variable clinical presentation, diagnostic features, follow-up and genetic analysis of four cases. Focal lobular spermatogenesis and pubertal acceleration associated with ipsilateral Leydig cell hyperplasia. Gonadotropin-independent familial sexual precocity with premature Leydig and germinal cell maturation (familial testotoxicosis): effects of a potent luteinizing hormone-releasing factor agonist and medroxyprogesterone acetate therapy in four cases. Mutations of gonadotrophin and gonadotrophin receptor genes: what do they teach us about reproductive physiology Activating mutations in the luteinizing hormone receptor gene: a human model of nonfollicle-stimulating hormone-dependent inhibin production and germ cell maturation. Pituitary gonadotropinindependent male-limited autosomal dominant sexual precocity in nine generations: familial testotoxicosis. Response to challenge with gonadotropin-releasing hormone agonist in a mother and her two sons with a constitutively activating mutation of the luteinizing hormone receptor-a clinical research center study. A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. The role of inhibins B and antimullerian hormone for diagnosis and follow-up of granulosa cell tumors. Aromatase p450 expression in a feminizing adrenal adenoma presenting as isosexual precocious puberty. Severe endocrine and nonendocrine manifestations of the McCune-Albright syndrome associated with activating mutations of stimulatory G protein Gs. Activating Gsalpha mutations: analysis of 113 patients with signs of McCune-Albright syndrome-a European Collaborative Study. Parental origin of Gsalpha mutations in the McCune-Albright syndrome and in isolated endocrine tumors. Sertoli tumors of the ovary: a clinicopathologic study of 28 cases with ultrastructural observations. Fracture incidence in polyostotic fibrous dysplasia and the McCune-Albright syndrome. Is McCune-Albright syndrome overlooked in subjects with fibrous dysplasia of bone Tamoxifen treatment for precocious puberty in McCune-Albright syndrome: a multicenter trial. Characterization and management of testicular pathology in McCune-Albright syndrome. Macroorchidism due to autonomous hyperfunction of Sertoli cells and G(s)alpha gene mutation: an unusual expression of McCune-Albright syndrome in a prepubertal boy. Testicular microlithiasis: an unreported feature of McCune-Albright syndrome in males. Polycystic ovaries, precocious puberty and acquired hypothyroidism: the Van Wyk and Grumbach syndrome. A potential novel mechanism for precocious puberty in juvenile hypothyroidism [see comments]. Naturally occurring mutations of the luteinizing-hormone receptor: lessons learned about reproductive physiology and G protein-coupled receptors. Genetic heterogeneity of constitutively activating mutations of the human luteinizing hormone receptor in familial male-limited precocious puberty. Luteinizing hormone receptor mutations in disorders of sexual development and cancer. Adult height after ketoconazole treatment in patients with familial male-limited precocious puberty. Treatment of familial male precocious puberty with spironolactone and testolactone. Long-term follow-up of spontaneous development in a boy with familial male precocious puberty. Nodular Leydig cell hyperplasia in a boy with familial male-limited precocious puberty. Concurrent hormone resistance (pseudohypoparathyroidism type Ia) and hormone independence (testotoxicosis) caused by a unique mutation in the G alpha s gene. A genome-wide association study of men with symptoms of testicular dysgenesis syndrome and its network biology interpretation. Exposure to exogenous estrogens in food: possible impact on human development and health. Natural history and incidence of premature thelarche in Puerto Rican girls aged 6 months to 8 years diagnosed between 1990 and 1995. Natural history of premature thelarche in Olmsted County, Minnesota, 1940 to 1984. Age at menarche and Tanner stage in girls exposed in utero and postnatally to polybrominated biphenyl. High incidence of central precocious puberty in a bounded geographic area of northwest Tuscany: an estrogen disrupter epidemic Urinary phthalates from 168 girls and boys measured twice a year during a 5-year period: associations with adrenal androgen levels and puberty. Serum hormones in boys prenatally exposed to polychlorinated biphenyls and dibenzofurans. Sonographic assessment of uterine and ovarian development in normal girls aged 1 to 12 years. Evaluation of pelvic ultrasonography in the diagnosis and differentiation of various forms of sexual precocity in girls. Pelvic ultrasonography in the evaluation of central precocious puberty: comparison with leuprolide stimulation test. Utility of breast ultrasonography in the diagnostic work-up of precocious puberty and proposal of a prognostic index for identifying girls with rapidly progressive central precocious puberty. Estimating volumes of the pituitary gland from T1-weighted magnetic-resonance images: effects of age, puberty, testosterone, and estradiol. Empty sella in children and adolescents with possible hypothalamic-pituitary disorders.
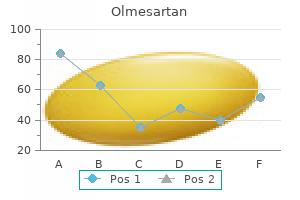
Proven 10mg olmesartan
The activity of these proteins correlates with active calcium transport; however blood pressure normal teenager generic olmesartan 20mg visa, a causal relationship remains to be established. Arrows indicate that the hexamers found in the upregulated rat osteocalcin gene are variants of a consensus sequence, repeated here with identical orientations (direct repeats). The best studied effect of vitamin D on the enterocyte is the induction of synthesis of the intestinal calcium-binding protein, calbindin-9K. The affinity of calbindin for calcium is approximately four times that of the brush border calcium-binding components, so calcium is preferentially transferred to calbindin. Calbindin serves to buffer the intracellular free calcium concentration during calcium absorption. It associates with microtubules and may play a role in the transport of calcium across the enterocyte. Vitamin D administration has been shown to increase the concentration of troponin C, a calcium-binding protein in muscle that plays a role in excitation coupling and increases the rate of uptake of calcium by the sarcoplasmic reticulum. The myopathy that accompanies vitamin D deficiency is characterized by normal creatine phosphokinase levels, a myopathic electromyogram, and biopsy findings of loss of myofibrils, fatty infiltration, and interstitial fibrosis. The myopathy resolves within days to weeks of vitamin D replacement and does not correlate with normalization of mineral ion homeostasis. Several analogues have been shown to have antiproliferative effects on normal cells as well as on malignant cells in vitro and in xenografts in immunosuppressed mice. This suggests that such analogues may be useful in the prevention and treatment of hyperparathyroidism. The antiproliferative effects of vitamin D have been exploited clinically in the treatment of psoriasis. The physiology underlying the differential biologic effects of these analogues is not completely understood. In contrast, the concentrations of these mineral ions in extracellular fluid are quite comparable. Extracellular calcium and phosphate, in particular, exist so close to the limits of their mutual solubility that stringent regulation of their concentrations is required to avoid diffuse precipitation of calcium phosphate crystals in tissues. Serum concentrations and total body balances of the mineral ions are maintained within narrow limits by powerful, interactive homeostatic mechanisms. In contrast, the mechanisms of the phosphate sensing needed for normal homeostasis are not understood. Dietary calcium restriction, for example, is followed by an increase in the efficiency of intestinal calcium absorption. Enhanced intestinal calcium absorption is quantitatively the most important response to calcium deprivation, but a series of other homeostatic events also occur that limit the impact of this stress. Major homeostatic responses to dietary calcium deprivation or loading are depicted. Arrow thickness indicates relative activity of transport or secretory mechanisms, whereas amounts of hormones or transported ions are related to the size of their notations. Note that the extracellular calcium concentration is well maintained, although different underlying mechanisms are involved in the two circumstances (see text for details). The concomitant increase in net bone resorption causes release of phosphate as well as calcium into the extracellular fluid. Moreover, nonenteral sources of calcium, such as intravenous calcium infusion or excessive net bone resorption (as from immobilization or malignancy), may readily overwhelm the limited homeostatic adaptations that remain once suppressed intestinal calcium absorption is bypassed. In such situations, the kidney rather than the intestine becomes the principal defense against hypercalcemia, and calcium homeostasis becomes critically dependent on adequate renal function. If renal function is impaired in these settings, as frequently occurs clinically, severe hypercalcemia and pathologic calcium deposition in extraskeletal sites may ensue. Although it seems plausible that such assays might prove particularly useful in some clinical situations, their role is presently unclear. They offer no advantage over older two-site assays, for example, in diagnosing primary hyperparathyroidism. Note some overlap between normal people and patients with primary hyperparathyroidism, but no overlap between hypercalcemic patients with primary hyperparathyroidism and those with hypercalcemia of malignancy. Advances in techniques for measurement of parathyroid hormone: current applications in clinical medicine and directions for future research. The measurements are based on single or double antibody radioimmunoassays or enzyme immunoassays, several of which are sufficiently sensitive to detect calcitonin deficiency. However, the double antibody assays are thought to provide the same information with less sample manipulation. The only clinical use of the calcitonin assay is as a tumor marker, primarily in medullary carcinoma of the thyroid. Because the assays measure both protein-bound and unbound vitamin D metabolites, results may not always reflect the levels of biologically relevant ("free") metabolites. This limitation may lead to misleading results in patients with nephrotic syndrome and vitamin D intoxication. With the move away from using radioligand-based assays, other methods for measuring vitamin D metabolites, including chemiluminescent assays, have been pioneered. Two normocalcemic patients with cancer (filled triangles) subsequently became hypercalcemic. The National Institute of Standards and Technology in the United States has developed standard reference materials for this purpose. Measurement of this metabolite should, therefore, be performed when vitamin D deficiency is suspected. Impaired 1-hydroxylation can contribute to the hypocalcemia of patients with renal dysfunction, oncogenic osteomalacia, and hereditary defects of vitamin D metabolism (see "Hypocalcemic Disorders"). An assay for the intact hormone is a classic sandwich assay with antibodies directed against both the N- and C-terminus of the hormone. Primary hyperparathyroidism results most often (75-80%) from the occurrence of one or more adenomas in previously normal parathyroid glands, although in 20% of cases diffuse hyperplasia of all parathyroid glands may be present or, rarely, parathyroid carcinoma may be found (less than 1-2%). The bone disease "osteitis fibrosa cystica" first was described by von Recklinghausen in 1891, but the etiologic link between this disease and parathyroid neoplasms was not established until 1925, when Mandl observed clinical improvement following removal of a parathyroid adenoma from a young male with severe bone disease. In early clinical descriptions of primary hyperparathyroidism, the disease emerged as a distinctly uncommon disorder with significant morbidity and mortality rates, in which nearly all affected patients manifested radiographically significant or symptomatic skeletal or renal involvement, or both. The skeletal involvement in "classical" primary hyperparathyroidism reflects a striking and generalized increase in osteoclastic bone resorption, which is accompanied by fibrovascular marrow replacement and increased osteoblastic activity. Note the dramatic remodeling associated with the intense region of high bone turnover in the third metacarpal in addition to widespread evidence of subperiosteal, endosteal, and trabecular resorption. The skull may exhibit a finely mottled, "salt-andpepper" radiographic appearance, with loss of definition of the inner and outer cortices. Dental radiographs typically show erosion or disappearance of the lamina dura due to subperiosteal resorption, often with extension into the adjacent mandibular bone. The erosion and demineralization of cortical bone may lead to radiographic disappearance of some bones, most notably the tufts of the distal phalanges of the hands, the inferolateral cortex of the distal third of the clavicles, the distal ulna, the inferior margin of the femoral neck and pubis, and the medial aspect of the proximal tibia. The clinical correlates of these changes may include aching bone pain and tenderness, "bowing" of the shoulders, kyphosis and loss of height, and collapse of lateral ribs and pelvis with "pigeon breast" and triradiate deformities, respectively. The renal manifestations of classical severe primary hyperparathyroidism include recurrent calcium nephrolithiasis, nephrocalcinosis, and renal functional abnormalities that range from impaired concentrating ability to end-stage renal failure. Associated signs and symptoms include recurrent flank pain, polyuria, and polydipsia. No unique features of the stone disease in primary hyperparathyroidism serve to distinguish it from that associated with other, more common causes of calcium kidney stones. The stone disease more often may be recurrent and severe, and in some patients, the stones may be composed entirely of calcium phosphate, instead of the pure oxalate or mixtures of oxalate and phosphate more commonly encountered in other disorders. In patients diagnosed before 1965, the frequency with which nephrolithiasis complicated primary hyperparathyroidism was as high as 60% to 80% (the frequency is currently less than 25%), yet in studies of unselected patients conducted throughout the past 50 years, primary hyperparathyroidism has accounted for fewer than 5% of all calcium kidney stones. Other clinical features that have been reported in association with classical severe primary hyperparathyroidism are conjunctival calcifications, band keratopathy, hypertension (50%), gastrointestinal signs and symptoms (anorexia, nausea, vomiting, constipation, or abdominal pain), peptic ulcer disease, and acute or chronic pancreatitis. The issue of whether primary hyperparathyroidism increases the risk for peptic ulcer disease and pancreatitis remains controversial. Although hyperparathyroidism is associated with a higher risk of hypertension, successful parathyroidectomy has not been shown to correct the hypertension. Signs and symptoms in primary hyperparathyroidism may result from the involvement of bone (fracture, bone pain) or kidneys (renal colic, renal failure), peptic ulcer disease, pancreatitis, or hypercalcemia per se (weakness, apathy, depression, polyuria, constipation, coma).
Cheap 40 mg olmesartan with visa
Effects of autonomic neuropathy associated with diabetes mellitus on cardiovascular function blood pressure ratio buy generic olmesartan 40 mg on-line. Primary nociceptive afferents mediate the blood flow dysfunction in non-glabrous (hairy) skin of type 2 diabetes: a new model for the pathogenesis of microvascular dysfunction. The effect of alpha-lipoic acid on the neurovascular reflex arc in patients with diabetic neuropathy assessed by capillary microscopy. Incidence rates and treatment of neuropathic pain conditions in the general population. Autoantibodies to autonomic nerves associated with cardiac and peripheral autonomic neuropathy. Long-term effect of converting enzyme inhibition on circadian sympathetic and parasympathetic modulation in patients with diabetic autonomic neuropathy. Pancreas transplantation restores epinephrine response and symptom recognition during hypoglycemia in patients with long-standing type I diabetes and autonomic neuropathy. Effects of glycemic control on heart rate variability in type I diabetic patients with cardiac autonomic neuropathy. Hypovolemia contributes to the pathogenesis of orthostatic hypotension in patients with diabetes mellitus. Efficacy of compression of different capacitance beds in the amelioration of orthostatic hypotension. Gastrointestinal motor dysfunction, symptoms, and neuropathy in noninsulin-dependent (type 2) diabetes mellitus. Cisapride versus placebo for 8 weeks on glycemic control and gastric emptying in insulin-dependent diabetes: a double blind cross-over trial. Domperidone in the management of symptoms of diabetic gastroparesis: efficacy, tolerability, and quality-of-life outcomes in a multicenter controlled trial. Comparison of metoclopramide and erythromycin in the treatment of diabetic gastroparesis. Sildenafil for treatment of erectile dysfunction in men with diabetes: a randomized controlled trial. Increased intimal-medial thickness in newly detected type 2 diabetes: risk factors. Postprandial plasma glucose is an independent risk factor for increased carotid intima-media thickness in non-diabetic individuals. Report of the National Heart, Lung, and Blood Institute/ American Heart Association Conference on Scientific Issues Related to Definition. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Metabolic syndrome with and without C-reactive protein as a predictor of coronary heart disease and diabetes in the West of Scotland Coronary Prevention Study. Risk for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome. An American Heart Association/ National Heart, Lung, and Blood Institute scientific statement. Cardiovascular events and correlates in the Veterans Affairs Diabetes Feasibility Trial. Congestive heart failure and cardiovascular death in patients with prediabetes and type 2 diabetes given thiazolidinediones: a meta-analysis of randomised clinical trials. Panel recommends easing restrictions on rosiglitazone despite concerns about cardiovascular safety. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. Glucagon-like peptide-1 receptor agonists for diabetes mellitus: a role in cardiovascular disease. Intensive glucose-lowering therapy reduces cardiovascular disease events in veterans affairs diabetes trial participants with lower calcified coronary atherosclerosis. Prevention of cardiovascular events and death in pravastatin patients with coronary heart disease and a broad range of initial cholesterol levels. Reduced coronary events in simvastatin-treated patients with coronary heart disease and diabetes or impaired glucose levels. Influence of low highdensity lipoprotein cholesterol and elevated triglyceride on coronary heart disease events and response to simvastatin therapy in 4S. Reduction of low-density lipoprotein cholesterol in patients with coronary heart disease and metabolic syndrome: analysis of the Treating to New Targets study. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of highdensity lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. Should the "high-intensity cholesterol-lowering therapy" strategy replace the "high-intensity statin therapy" The role of niacin in raising highdensity lipoprotein cholesterol to reduce cardiovascular events in patients with atherosclerotic cardiovascular disease and optimally treated low-density lipoprotein cholesterol, Rationale and study design. Determinants and importance of stress hyperglycaemia in non-diabetic patients with myocardial infarction. The prognostic value of blood glucose in diabetic patients with acute myocardial infarction. Stress hyperglycemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Sulfonylurea drugs increase early mortality in patients with diabetes mellitus after direct angioplasty for acute myocardial infarction. Oral sulfonylurea hypoglycemic agents prevent ischemic preconditioning in human myocardium: two paradoxes revisited. Coronary vascular responsiveness to adenosine is impaired additively by blockade of nitric oxide synthesis and a sulfonylurea. Improvement in endothelial function by angiotensin-converting enzyme inhibition in noninsulin-dependent diabetes mellitus. Effects of ramipril on plasma fibrinolytic balance in patients with acute anterior myocardial infarction. Effect of the angiotensinconverting enzyme inhibitor trandolapril on mortality and morbidity in diabetic patients with left ventricular dysfunction after acute myocardial infarction. Diuretics and -blockers do not have adverse effects at 1 year on plasma lipid and lipoprotein profiles in men with hypertension. Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. Antihypertensives and the risk of serious hypoglycemia in older persons using insulin or sulfonylureas. Metabolic and cardiovascular effects of carvedilol and atenolol in non-insulin-dependent diabetes mellitus and hypertension. Drug resistance and pseudoresistance: an unintended consequence of enteric coating aspirin. Effects of an angiotensin-converting enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. Clinical outcomes in antihypertensive treatment of type 2 diabetes, impaired fasting glucose concentration, and normoglycemia. Acute myocardial infarction in the diabetic patient: pathophysiology, clinical course and prognosis. Impaired circadian modulation of sympathovagal activity in diabetes: a possible explanation for altered temporal onset of cardiovascular disease. Effect of autonomic nervous system dysfunction on the circadian pattern of myocardial ischemia in diabetes mellitus. Plasma fibrinogen-a new factor of the metabolic syndrome: a population-based study. Increased plasminogen activator inhibitor type 1 in coronary artery atherectomy specimens from type 2 diabetic compared with nondiabetic patients: a potential factor predisposing to thrombosis and its persistence. Influence of diabetes mellitus on clinical outcome in the thrombolytic era of acute myocardial infarction. Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries.
Olmesartan 10mg generic
Furthermore blood pressure understanding purchase olmesartan 20mg, measures of fasting and postchallenge glucose concentrations in the same individual over time are less reproducible than those for the HbA1c. In the absence of unequivocal hyperglycemia, a diagnostic result should be confirmed by repeat testing. If test results are normal, repeat testing should be carried out at 3- to 5-yr intervals. Age at initiation and frequency of screening to detect type 2 diabetes: a cost-effectiveness analysis. In addition, affected individuals have a greater likelihood of having dyslipidemia, hypertension, and obesity. Therefore, it is important for the clinician to screen for diabetes in a cost-effective manner in subjects who demonstrate major risk factors for diabetes as summarized in Table 31-3. A number of environmental factors have been shown to play a critical role in the development of the disease, particularly excessive caloric intake leading to obesity and a sedentary lifestyle. The clinical presentation is also heterogeneous, with a wide range in age at onset, severity of associated hyperglycemia, and degree of obesity. The most common factors that place an increased secretory burden on the beta cell are puberty, pregnancy, a sedentary lifestyle, and overeating leading to weight gain. An underlying genetic predisposition appears to be a critical factor in determining the frequency with which beta cell failure occurs. The genes involved in the common polygenic forms of the disorder have been far more difficult to identify and characterize. Monogenic Forms of Diabetes In the monogenic forms of diabetes, the gene involved is both necessary and sufficient to cause disease. In other words, environmental factors play little or no role in determining whether a genetically predisposed person develops clinical diabetes. The monogenic forms of diabetes usually are diagnosed in younger patients, often in the first 2 to 3 decades of life; however, if only mild, asymptomatic elevations in blood glucose occur, the diagnosis may be missed until later in life. The monogenic forms of diabetes are summarized in Table 31-5 and can be divided into those in which the mechanism is a defect in insulin secretion and those that involve defective responses to insulin or insulin resistance. Numerous mutations have been identified in the insulin receptor gene in various insulin-resistant patients. Type A insulin resistance is defined by the presence of insulin resistance, acanthosis nigricans, and hyperandrogenism. The intrinsic function of the receptor may be abnormal if the affinity of insulin binding is reduced (class 3) or if receptor tyrosine kinase is inactivated (class 4). The insulin resistance that is associated with insulin receptor mutations can be severe, manifesting in the neonatal period. In another monogenic form of diabetes, lipoatrophic diabetes, severe insulin resistance is associated with lipoatrophy and lipodystrophy. This form of diabetes is characterized by a paucity of fat, insulin resistance, and hypertriglyceridemia. People homozygous for the Pro12 allele are more insulin resistant than those with one Ala12 allele and have a 1. There is also evidence for interaction between this polymorphism and fatty acids, linking this locus with diet. A second polymorphism, C161 T, has been linked to insulin resistance in Hispanic and non-Hispanic white women. The prevalence of all causes of neonatal diabetes has been estimated to be between 1 in 100,000 and 1 in 300,000 live births. If the onset is before 6 months of age, a genetic reason is the most likely underlying cause. The presence of hyperglycemia is often undetected, and the diagnosis is then made when the clinical condition deteriorates due to marked hyperglycemia with or without ketoacidosis. Associated features include low birth weight below the 10th percentile (especially in the absence of maternal diabetes), developmental delay, learning disorders, speech disorders, muscle weakness especially with climbing stairs, and seizures. Occasionally, multiple family members are also found to have early-onset, relapsing, or nonobese young adult appearance of diabetes, but most cases are sporadic. If the channel does not close at physiologic levels of glucose, hypoinsulinism and hyperglycemia result. This causes severe diabetes with low or negative C-peptide and ketosis in the first few weeks of life. Mutations in these genes that cause the opposite condition, decreased channel function, are a cause of familial hyperinsulinemia with hypoglycemia. However, it is critical that this be done after a mutation has been documented, because the protocol involves high doses of sulfonylureas (administered in divided doses and off-label in the United States for children) and simultaneous aggressive insulin withdrawal. Collaboration with or referral to a center with experience in this treatment is highly encouraged because of potential side effects and other adverse effects. In the case of unaffected parents with one affected child, several studies have reported germline mosaicism as a known or possible cause of the presence of the syndrome in several siblings in one family. Therefore, the risk that each subsequent child will have neonatal diabetes can range from less than 10% to 50%, depending on the presence of mosaicism in the gametes. Affected persons characteristically have marked hyperinsulinemia on routine insulin assays. Increases in the concentration of insulin in association with diabetes usually indicate insulin resistance, but in this syndrome, insulin resistance can be easily excluded because the patients respond normally to administration of exogenous insulin. The increased concentrations of insulin appear to be related to the presence of mutations in regions of the insulin molecule that are important for receptor binding, particularly the carboxy-terminus of the insulin B chain. Because the liver is the major site of insulin clearance and first-pass hepatic insulin uptake and degradation are mediated by the insulin receptor, mutant forms of insulin with diminished insulin receptor binding ability are cleared more slowly from the circulation, and this reduction in insulin clearance leads to hyperinsulinemia. Alternatively, mutations in proinsulin can reduce the conversion of proinsulin to insulin, leading to accumulation of proinsulin. A patient with a mutation in prohormone convertase 1, one of the enzymes responsible for the conversion of proinsulin to insulin, has been described. The mitochondrion plays a key role in the regulation of insulin secretion, particularly in response to glucose. Abnormal insulin secretion may be seen in subjects with this mitochondrial mutation, even if diabetes has not yet developed and glucose tolerance is normal or impaired. Prospective testing indicates that in most patients the disease onset occurs in childhood or adolescence. In some patients, there may be a rapid progression to overt asymptomatic or symptomatic hyperglycemia, necessitating therapy with an oral hypoglycemic drug or insulin. All the susceptibility genes identified to date cause impaired insulin secretory responses to glucose, although the mechanisms differ. Glucokinase is expressed at its highest levels in the pancreatic beta cell and the liver. Glucokinase functions as the glucose sensor in the beta cell by controlling the rate of entry of glucose into the glycolytic pathway (glucose phosphorylation) and its subsequent metabolism. In the liver, glucokinase plays a key role in the ability to store glucose as glycogen, particularly in the postprandial state. In the pancreatic beta cell, these transcription factors regulate the expression of the insulin gene as well as proteins involved in glucose transport and metabolism and mitochondrial metabolism (all linked to insulin secretion) and lipoprotein metabolism. Persons with diabetes related to mutations in these genes have defects in insulin secretory responses to a variety of secretagogues, particularly glucose, that are present before the onset of hyperglycemia, suggesting that they represent the primary functional defect in the syndrome. Reduced glucagon responses to arginine have also been observed, suggesting that the pancreatic alpha cell is also involved in a broader pancreatic developmental abnormality. It also plays a central role in the development of the pancreas and in regulation of the expression of a variety of pancreatic islet genes, including (besides insulin) the genes encoding glucokinase, islet amyloid polypeptide, and glucose transporter 2. Earlier genetic studies relied either on the candidate gene approach, in which the search for diabetes genes was dictated by the prevailing understanding of the pathways involved in glucose regulation, or on linkage studies. Parents are genotyped at a particular marker, and the offspring are scored for sharing of zero, one, or two alleles inherited from their parents. Markers are genotyped in family members in the regions of polymorphic repeats called microsatellites or simple tandem repeats. The development of this approach depended on a number of factors including the completion of the Human Genome Project, the genotyping of 3. This is a rapidly changing field, and it is certain that the list of diabetes genes will increase. However, two meta-analyses of all the published data supported a role for this gene in diabetes susceptibility.
Buy generic olmesartan 10mg line
Inhaled technosphere insulin: a novel delivery system and formulation for the treatment of types 1 and 2 diabetes mellitus blood pressure natural order olmesartan 10mg otc. Review of pramlintide as adjunctive therapy in treatment of type 1 and type 2 diabetes. However, we have elected to begin this chapter by building on the story of a German pathologist named Martin Schmidt who, in 1902, noted a small peri-islet cellular infiltrate on microscopic evaluation of the pancreas obtained at autopsy from a 10-year-old child with diabetes. Works shortly thereafter by Shields Warren in the 1920s drew attention to the relationship between this infiltrate and the age of diabetes onset. Further contributing to the overall setting of diagnostic complexity has been movement to increasingly diverse genetic admixtures due to geographic migration and social changes. This said, as for most animal models of disease, only a limited number of therapeutic efforts have been effectively translated to humans, whereas in research settings one observes a clear clinical benefit. Very different Type1Diabetes Autoimmune disorder marked by destruction of insulin-producing beta cells and loss of insulin production Rapid onset; very high to extremely high blood glucose levels; polyphagia; polydipsia, polyurea; ketoacidosis Sudden (symptoms for days to weeks) Family history of autoimmune disease but in particular, T1D (10-fold increased risk versus general population) Typically early life through adolescence but can occur at any age Absolute requirement for insulin (multiple daily injections or insulin pump); self-management lifestyle modification (monitor food types, exercise, etc. Complications Mostly similar, but some variation Acute emergencies of hypoglycemia and ketoacidosis leading to hypoglycemic unawareness; chronic effects of hyperglycemia can lead to retinopathy, nephropathy, neuropathy, cardiovascular disease, etc. Interestingly, with respect to T cells, a relative overrepresentation appears with respect to the number of cells bearing a T-cell receptor specific for a given beta-cell antigen, namely, insulin. Studies of islet beta-cell mass indicate that both destruction and replication/regeneration are present months before the onset of diabetes,51 although there is convincing evidence of an acceleration of beta-cell destruction at disease onset. However, recent convincing evidence exists to suggest that in terms of beta-cell replication, rodent models differ dramatically from human physiology, with beta-cell replication forming a rare event in humans from adolescence through adulthood. Hence, these molecules play a key role in controlling overall immune responses including those autoimmune in nature that result in beta-cell destruction. In low doses, a more chronic diabetes (hyperglycemia) develops that is likely to have some immunologic derivation, meaning pathogenesis tied to immune responses against beta cells. Other examples of compounds used to induce diabetes include the toxin alloxan as well as viruses that target the pancreas, including islet cells. These same studies also noted that Induced Models of Type 1 Diabetes Mellitus Diabetes, and even insulitis, can be induced in several strains of rodents by means of drugs that induce islet cell destruction and broadly activate immune responses, including those directed against beta-cell antigens. Histopathologic examination of the same section of pancreas showed that a normal islet with no immune infiltrate can coexist with an islet containing beta cells with intense infiltration as well as a pseudoatrophic islet that has no infiltrate. This spottiness of the pathologic process is reminiscent of the destruction of areas of the skin in patients with vitiligo, in which melanocytes are destroyed in patches. Molecules such as Fas may be important as T cells expressing Fas ligand can induce apoptosis of beta cells. This conclusion is based on a series of observations including the age at disease onset, form of symptomatic presentation. Overview of Prevalence and Genetics In the United States, the risk of childhood diabetes is approximately 1 in 300. B, Time-based trends for the incidence of T1D in children ages 0 to 14 years in areas with high or high-intermediate rates of disease. For a locus that contributes to disease in a recessive manner, only one quarter of dizygotic twins would be homozygous to a sibling with diabetes at that locus, but all monozygotic twins would be homozygous for all recessive loci of their diabetic twin. With such genetic heterogeneity, one would expect different concordance rates for different genetic causes. Redondo and coworkers141 analyzed prospective followup data from a large series of initially discordant monozygotic twins from Great Britain combined with a series from the United States. Nevertheless, the hazard rate for development of diabetes decreased as the period of discordance increased. There was also a marked variation in the risk of diabetes relative to the age at which the disorder developed in the index twin. With long-term follow-up, the overall rate of concordance for monozygotic twins exceeds 50%. If diabetes developed in the index twin before age 5 years, the concordance rate was 70% after 40 years of follow-up. If the risk is identical, it suggests that environmental factors whose presence is time-dependent. Dizygotic twins differ from siblings in terms of a greater commonality of environment over time. Interestingly, studies of dizygotic twins suggest that their risk of diabetes may not differ from that of siblings or, at most, may be increased by a factor of two compared with the 10-fold increase for monozygotic twins. Each molecule is made up of two chains, and each chain is encoded by a separate gene. Likewise, for the class I molecules (A, B, and C), only a single chain is specified, because the other chain, 2-microglobulin, is minimally polymorphic. When specific alleles of different genes are nonrandomly associated with each other on a haplotype. Linkage disequilibrium is not the same as linkage, although to have linkage disequilibrium, genes must be linked. Genes are linked when they are close together on the same chromosome and thus transmitted from parent to child as a haplotype group. If alleles of linked genes are nonrandomly associated with each other in a population, they are in linkage disequilibrium. This adds an important level of diversity in that the protein chains encoded by the alleles of one haplotype can combine with the chains encoded by the other haplotype. The variant associated with disease risk results in gain of function and decreased T-cell receptor signaling. Environmental factors (discussed later) also play a part in the development of polygenic forms of diabetes. However, additional and rare forms of diabetes, termed monogenic, result from mutations in a single gene. Monogenic forms of diabetes account for about 1% to 5% of all cases of diabetes in young individuals. In most cases of monogenic diabetes, the gene mutation is inherited; in the remaining cases the gene mutation develops spontaneously. GeneorSyndrome TypicalAge atOnset Typeof Inheritance orMutation Transientor Permanent If genetic testing is not performed, people with monogenic diabetes may appear to have one of the polygenic forms of diabetes. Importantly, some monogenic forms of diabetes can be treated with oral diabetes medications, whereas other forms require insulin injections. A correct diagnosis that allows the proper treatment to be selected should lead to better glucose control and improved health in the long term. This notion becomes relevant in that it has been hypothesized that greater expression of insulin and other tissue-specific antigens leads to tolerance and disease suppression. It is associated with insulitis and beta-cell destruction as well as lymphocytic intestinal inflammation with flattened villi and severe malabsorption. It is inherited as an X-linked recessive disease affecting only boys, with a frequent clinical history of lack of male births. This is an important syndrome to recognize early as bone marrow transplantation, affording restoration of functional T-regulatory cells (even with partial chimerism), has proved to be therapeutic. Following, some infants demonstrate protracted growth and weight gain, but appropriate therapy has the ability to improve and may in fact normalize growth and development. Consistent with this conclusion are the regional differences in disease rates based on geography, seasonality in its diagnosis, rising incidence trends, and variance among twins. However, despite decades of research, no single environmental agent has been identified that would universally explain these observations. Factors that increase diabetes risk may be increasing, or just as likely, factors that suppress the development of diabetes may be decreasing. In terms of this rising incidence, it is quite interesting that it is occurring at different rates globally as a function of age at onset. The accelerator and overload hypotheses suggest that environmental stresses, specifically childhood obesity for the former, increase insulin demand, thereby overloading the islet cells and accelerating beta-cell autoimmune damage. However, as a collective, most fall into the groups involving infection (viruses in particular), vaccination, and diet. Initial anecdotal reports that coxsackievirus (a form of enterovirus) infections might cause diabetes involved children who had severe infections and who died at disease onset. At the time of presentation with diabetes, almost all children have a rising elevation in HbA1c, reflecting what is likely months of hyperglycemia preceding diagnosis. Enteroviral infection was found to be associated with the appearance of anti-islet autoantibodies in some regions (Finland)195 but not in others (Colorado),196 a finding that may be related to the frequency and timing of enteroviral infections in these two populations.
