

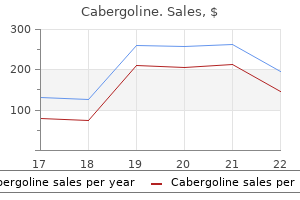
0.25mg cabergoline mastercard
Ataxia may be severe menstruation and breastfeeding order on line cabergoline, preventing an affected patient from standing without assistance. Fingertonose and heeltoshin tests are often normal when the patient is tested in the bed. Truncal ataxia frequently becomes obvious only on standing or sitting, reflecting midline degeneration of the superior division of the vermis. It presents with slowly progressive muscular weakness, sensory impairment, and hyporeflexia, accompanied by burning feet and lancinating pains. The classic clinical triad of ataxia, changes of consciousness, and oculomotor abnormalities is present in only a minority of patients. Clinical presentation is usually subtle, especially in nonalcoholic patients and in those with deep coma whose neurological evaluation is limited. Some individuals are at increased risk because of a genetic predisposition, which means that their thiamine requirements are increased and higher blood concentrations are required for thiamine to enter the brain cells. Patients with thiamine deficiency may become acutely symptomatic when challenged with large doses of carbohydrate. Changes to thiamine transport and utilization mean that higher doses of thiamine are required to treat the condition successfully in alcohol misusers, and that the thiamine must be given parenterally. This should be accompanied by magnesium and fluid replacement, according to separate guidelines from the Royal College of Physicians (2001), British Association for Psychopharmacology (2004), European Federation of Neurological Societies (2010), and National Institute for Health and Care Excellence (2011). The dry form of neuropathy is characterized by symmetric impairment and hyporeflexia, most marked in the distal segments of the limbs. Dysesthesias are prominent with a dull, constant ache in the feet and legs, or lightning pains. Cramps, bandlike feelings, coldness of the feet, and burning in the soles are also common, and are worsened by contact with bedclothes or the ground (burning feet). The signs are those of sensorymotor neuropathy, initially with distal weakness and predominant cutaneous sensory loss. Nerve conduction study and electromyography generally reveal a moderate decrease of motor and sensorimotor nerve velocity. The wet form manifestations include severe leg edema, heart murmur, accentuated secondary pulmonary sounds, and cardiac enlargement, together with a severe neuropathy, predominantly affecting distal motor function. Serum level and urinary excretion of thiamine are reduced, while lactate in serum is increased. The salient features include anterograde amnesia (impaired ability to acquire new information) and retrograde amnesia (impaired ability to recall events that has been well established before the onset of the syndrome). Immediate memory (digit repetition) and remote memory (early life events) are relatively unaffected. Confabulation (momentary and fantastic memory) is not constant and often is associated with false recognition. A collateral history from family or friends should be taken to determine the premorbid functioning state of the patient, alcohol use history and complications, and the observed decline in functioning. Assessment of cognitive function should not be carried out until at least 4 weeks post alcohol withdrawal. Pathologically, there are symmetric lesions in both mammillary bodies with necrosis, neuronal loss, and hemorrhages, and in the bilateral medial thalami. Cerebellar degeneration Thiamine deficiency probably plays a part in cerebellar degeneration, particularly in alcoholics. More frequent in men, this disorder is characterized by gait instability and ataxia. The pathological changes consist of degeneration of the cerebellar cortex, particularly of the Purkinje cells. The syndrome is caused by nutritional deficiency rather than the toxic effects of alcohol. Central pontine myelinolysis is related to a profound electrolytic disturbance, associated with hypoxia and vitamin B1 deficiency, also called osmotic demyelinization syndrome. The nature of the lesions is essentially demyelination involving the basis pontis. Most patients who develop this syndrome have chronic medical illnesses that are treated by hemodialysis. Central pontine myelinolysis is induced by a rapid correction of hyponatremia, causing the extracellular fluid to be relatively hypertonic, with secondary damage to the pontine and extrapontine cells of the brain. Primary clinical manifestations consist of progressive gait disturbance, postural instability, hallucinations, and mild cognitive dysfunction, progressing to paraparesis or quadriparesis, dysphagia, dysarthria, diplopia, and loss of consciousness (lockedinlike syndrome) over a period of several days. Nicotinic acid/niacin deficiency Central pontine myelinolysis Polyneuropathy (neuropathic beriberi) the main pathological change in polyneuropathy is axonal degeneration with destruction of both axon and myelin sheath. The most pronounced changes are observed in the distal part of the largest myelinated fibers. Anterior horn and dorsal root ganglion cells undergo chromatolysis, indicating axonal damage. Clinically there are three forms of beriberi: dry beriberi, wet beriberi, and infantile beriberi. Dry beriberi is characterized by a sensorimotor, painful, distal, axonal peripheral neuropathy. Wet beriberi is associated with highoutput heart failure with peripheral neuropathy. The terms wet and dry beriberi have been used to describe the presence or absence of edema in neuropathic beriberi. Pellagra Pellagra, or rough skin, continues to occur in parts of Africa and Asia, especially in populations dependent on corn as the principal source of carbohydrate. In developed countries niacin deficiency is 406 Part 11 Specific Toxicities and Deficiencies seen in alcoholics and patients taking isoniazid. There is a single case report of a patient developing nicotinic acid deficiency after valproic acid and phenobarbital therapy. Pregnant women are protected from niacin deficiency owing to their enhanced ability to convert tryptophan to niacin endogenously, particularly during the third trimester. In human pellagra, neuropathological abnormalities consistently observed are chromatolysis in motor neurons (Betz cells in the motor cortex, brainstem nuclei, anterior horn cells of the spinal cord), characterized by cytoplasmic swelling, disappearance of Nissl granules, and displacement of flattened nucleus to the periphery of the cell body. Pellagra affects the skin, the gastrointestinal system, and the central nervous system. In industrialized countries, particularly among alcoholics, niacin deficiency may present only with encephalopathy. Patients may have altered sensorium, diffuse rigidity of the limbs, and grasping and sucking reflexes. Dementia and confusion are the most consistent findings, followed by diarrhea (50%) and dermatitis (about 30%). The initial symptoms can be insomnia, fatigue, nervousness, irritability, and depression. The detailed neurological examination may disclose psychomotor slowing, apathy, and memory impairment. Peripheral neuropathy is frequent and may be indistinguishable from the neuropathic beriberi. Coexisting deficiencies of thiamine and pyridoxine are common, especially in alcoholics. The aminoaciduria is attributed to the defective transporter in the kidney, but the impact of the transport defect on intestinal absorption and its connection to pellagra or neurological manifestations is still unclear. In the evaluation of potential niacin deficiency, nicotinic acid metabolites can be identified in the urine; however, clinical suspicion and general availability of niacin make this measurement impractical and unnecessary. With proper therapy, the prognosis for the resolution of neurological symptoms is excellent. Nicotinic acid deficiency encephalopathy in alcoholic patients In 1940, Jolliffe et al. Although endemic niacin deficiency has essentially been eradicated in most Western countries, alcohol withdrawal delirium may in many cases account for pellagra. There are several case reports of patients who misuse alcohol presenting with laryngitis and psychosis due to nicotinic acid deficiency. Chronic niacin deficiency Chronic niacin deficiency is a dramatic example of how nutritional deficiency can affect mental function in childhood.
Buy cabergoline 0.5 mg low cost
Mycobacterium avium has recently been observed to result in chronic pulmonary infections in otherwise healthy elderly persons breast cancer vector buy cheap cabergoline, and under these circumstances occasionally invades the brain with abscess formation. Diagnosis can only be achieved with specialized histological analysis and bacterial culture. Differential diagnosis includes cryptococcus, bartonella, cytomegalovirus, syphilis, and toxoplasmosis. Surgical removal combined with a combination of ethambutol, clarithromycin, and rifampicin has been effective in some cases, although side effects often form a considerable or absolute barrier to persistent antibiotic treatment. Tuberculosis of the central nervous system: Overview of neuroradiological findings. It affects mainly the skin, peripheral nerves, upper respiratory airways, anterior eye segments, and testes. Bacteriology detected during 2012 was 232,857 (excluding the small number of cases in Europe). Most countries that were previously highly endemic for leprosy have eliminated it at the national level. Leprosy today prevails in the poor areas of the world, particularly in the tropical regions. The most important source of leprosy is infected humans, but armadillos, chimpanzees, and monkeys are other reservoirs of leprosy. The carcasses of nonviable organisms in tissues or in smears can be detected by silver staining methods. Pathogenesis the two portals of entry seriously considered are the upper respiratory tract and the skin, in which transmission through secretions from the nasal mucosa of untreated patients is thought to be the main route of infection. Bacteremia is present in up 15% of paucibacillary patients and is common in multibacillary patients. Prevalence rates vary geographically, with 75% of leprosy occurring in Southern Asia, 12% in Africa, and 8% in the Americas. Clinical features Skin lesions with associated sensory loss and enlarged peripheral nerves are the cardinal symptoms and signs of leprosy. Often sensory abnormalities precede paralysis, with impaired temperature and touch frequently linked. Cutaneous nerves and superficial peripheral nerve trunks are often enlarged in the region of lesions. The commonly involved nerves that present indurated hypertrophy are the great auricular, ulnar, radial, fibular, and sural nerves. Acrodystrophy and autoamputation are the common late complications of neuropathy, in which loss of pain is predominant. Some regions such as scalp, palms, soles, and midline of the back are, however, not involved. Skin lesions include macules, nodules, papules, ulcerations, and diffuse myxedemalike involvement. Sensory loss is commonly first distributed to the ear helices, nose, malar regions, dorsal surface of the hands, forearms, feet, and dorsolateral surfaces of the lower legs. Other areas are the upper respiratory tract from the nasal mucosa to the larynx, the eye, lymph nodes, and testes. Commonly affected nerves are the ulnar, posterior tibial, common peroneal, and the median and facial nerves. The preservation of tendon reflexes is one particular aspect of the clinical findings of neuropathy associated with leprosy that should be mentioned, as this can help differentiate from lengthdependent neuropathies where tendon reflex loss is a hallmark. Indeterminate leprosy (I) usually presents as hypopigmented, or slightly erythematous, poorly defined macules. Texture, the amount of hair, sensation, and sweating in the affected area are, at the most, only slightly changed. Because of this vague and nonspecific feature, indeterminate lesions can be diagnosed only with close cooperation between clinician and pathologist. Obstructive vasculitis causes massive dermal infarcts, and ulcers can later supervene as this form of leprosy progresses. Same patient with facial paralysis on the right side, and macular lesions of the upper and middle right hemiface. Leprosy reactions Inflammatory immune reactions that occur during the progression and the treatment of leprosy should also be taken into account because of their harmful effects on the patient. There are two main reactions: type 1, or reversal reactions, and type 2, or erythema nodosum leprosum reactions (Table 65. Clinical findings include aggravation of previous skin lesions, new skin lesions, and neuritis, which usually appears during the first few months following initiation of chemotherapy. By way of repeated reversal reactions, borderline lesions may gradually change toward tuberculoid leprosy or tuberculoid lesions toward scar tissue. Permanent neurological deficits may result unless antiinflammatory treatment is quickly initiated. These type 2 reactions often arise after several months or more of therapy, but may also develop in untreated patients. Skin smears should be obtained from multiple sites, frequently at two sites, including the edges of macules or plaques, or nodules, and earlobes. Only in rare instances is there a need to use laboratory and other investigations to confirm a diagnosis of leprosy. A careful physical examination of the entire skin surface and superficial peripheral nerves to look for skin lesions, sensory changes, peripheral nerve enlargement, and motor deficit. Sensory changes are the most important criteria for clinical diagnosis of leprosy. A person presenting with skin lesions or with symptoms suggestive of nerve damage, in whom the cardinal signs are absent or doubtful, should be called a "suspect case" in the absence of any immediately obvious alternate diagnosis. Such individuals should be told the basic facts of leprosy and advised to return to the center if signs persist for more than six months or if at any time worsening is noticed. Suspect cases may be also sent to referral clinics that have more facilities for diagnosis. The paucibacillary regimen includes rifampicin 600 mg given once monthly under supervision, plus dapsone 100 mg daily for 6 months. The treatment regimen for single skin lesion paucibacillary leprosy includes rifampicin 600 mg, ofloxacin 400 mg, and minocycline 100 mg. The multibacillary regimen includes rifampicin 600 mg and clofazimine 300 mg given once monthly under supervision, plus dapsone 100 mg/day and clofazimine 50 mg/day for 12 months. Prothionamide, ethionamide, or minocycline may be used as a substitute for clofazimine when there is hyperpigmentation of the skin from clofazimine. Ofloxacin, clarithromycin, and minocycline are currently used in clinical trials and may be used as an alternative regimen when patients are allergic to rifampicin. Other investigations the lepromin test consists of the intradermal inoculation of 0. This skin test is never used as a diagnostic test of leprosy, as many people in the general population are reactive. Motor and sensory nerve conduction velocity studies can be helpful in demonstrating abnormality in the nerve trunks and branches, which often predates clinical characteristics. Treatment of leprosy reactions Differential diagnosis the differential diagnoses of leprosy are extensive and include sarcoidosis, syphilis, yaws, granuloma annulare, leishmaniasis, lupus erythematosus, superficial mycoses, lymphoma, psoriasis, pityriasis rosacea, neurofibromatosis, syringomyelia, lead toxicity, diabetes mellitus, primary amyloidosis, sensory polyneuropathies, other mononeuropathies, and familial hypertrophic neuropathy. Immobilization by splint, analgesics, and prednisone are the mainstay of the treatment of acute neuritis in reversal reaction. Longterm corticosteroid therapy may be given, preferably in an alternated day regimen.
Purchase genuine cabergoline line
Gait problems are typically accompanied by "frontal" cognitive and behavioral symptoms women's health clinic douglasville ga order cabergoline 0.5 mg free shipping, such as mental slowness, perseveration, decrease in verbal fluency, and dysphoria. Any normal voluntary movement is regarded as the result of precisely coordinated, "orchestrated" activity of a variety of antagonistic and synergistic muscles. This temporally and spatially ordered interplay between different muscles is realized through extensive bilateral connections of the cerebellum, with structures of the central nervous system participating in motor functions at different levels (motor cortex, basal ganglia, brainstem nuclei, reticular formation, spinal motor neurons, proprioceptive neurons, and pathways). The cerebellum as the main "motor coordinator" receives advance information about any changes of muscle tone and posture of body segments, as well as about any intended movement. Using this advance information, the cerebellum "prepares" the correct activity of voluntary muscles, organizes fine motor control, and ensures accurate execution of the movement. While under normal conditions cerebellar functional circuitry consisting of many feedback and feedforward connections is preserved, cerebellar disorders lead to malfunctioning of this circuitry and result in desynchronization of muscle contractions. Handicaps in cerebellar motor control clinically manifest as confused, irregular "jolts" leading to scanning speech, intention tremor, dysmetria, body titubation (a coarse foreandaft tremor of the trunk), and other cerebellar phenomena. The existence of some corticopontocerebellar projections representing the dorsal stream of cognitive processing suggest that the cerebellum also may play a role in spatial awareness and attentional functions. Cerebellar ataxic disorders the cerebellum and cerebellar pathways are affected in a variety of acute and chronic conditions that can cause ataxia (Table 55. In these disorders ataxia is frequently associated with headache, vomiting, vertigo, brainstem signs, and cranial nerve involvement. One should remember that even small cerebellar infarcts and hemorrhages are regarded, due to the limited volume of the posterior fossa, as potentially lifethreatening conditions in view of the high risk of obstructive hydrocephalus on the development of brain edema and remote pressure effects. The same is true for any other disorders characterized by large, acute cerebellar lesions with rapidly progressive edema of the posterior fossa structures. Because of the risk of herniation in these patients, lumbar puncture is strongly contraindicated. Repeated paroxysms of acute ataxic symptoms are seen in periodic (episodic) ataxias. These hereditary diseases are caused by genetic defects of calcium or potassium ion channels, which result in abnormalities of neuronal membrane excitability. Some patients with ataxic paroxysms may benefit from administration of acetazolamide (acetazolamideresponsive forms of periodic ataxias). Chronic ataxia Chronic ataxia may be caused by a number of different disorders (see Table 55. Chronic progressive ataxia is a key feature of idiopathic degenerative ataxic syndromes, both hereditary and sporadic. Other clinically important recessive ataxias include ataxiatelangiectasia, ataxia with oculomotor apraxia, several forms of spastic ataxia, and several forms of congenital nonprogressive cerebellar hypoplasia. Sporadic (idiopathic) degenerative ataxia Sporadic (idiopathic) degenerative ataxia is a heterogeneous entity comprising parenchymatous cortical cerebellar atrophy and olivopontocerebellar atrophy. The latter is now regarded as a form of multiple system atrophy, a severe neurodegenerative disorder characterized by involvement of many cerebral and spinal systems (cerebellum, basal ganglia, brainstem, spinal autonomic nuclei, and motor neurons) and the presence of specific asynucleinpositive glial cytoplasmic inclusions. Hereditary ataxias Hereditary ataxias are a clinically and genetically heterogeneous group of disorders transmitted, most frequently, as autosomal dominant or autosomal recessive traits; each of these major subtypes of hereditary ataxias also demonstrates striking genetic heterogeneity. There are nearly 70 distinct genetic forms of "pure" hereditary ataxias and nearly 300 additional genetic conditions manifesting, among others, by ataxic symptoms. The differential diagnosis between these conditions represents a serious challenge owing to significant phenotypic overlap. There is an inverse correlation between the copy number of trinucleotide repeats in the mutant gene and the age at disease onset. Moreover, the longest expanded alleles are associated with the most severe phenotypes. Recently, a new technology of exome sequencing made it possible to perform genomewide analysis for genetically heterogeneous disorders, such as hereditary ataxias, which may greatly facilitate the search for mutations in a variety of genes. In patients with sporadic ataxia, a detailed search is needed for possible somatic disorders that may cause cerebellar dysfunction (malignancies, endocrine and hepatic disorders, etc. Since ataxia may be a major presentation of a variety of metabolic diseases (see Table 55. Management Management and prognosis of ataxic syndromes rely on the primary cause of ataxia. If radical therapy is possible (such as surgery of cerebellar tumor or vitamin supplementation in specific vitamin deficiency), one can expect complete or partial recovery, or at least cessation of further disease progression. Limited positive effect has been reported in degenerative ataxias with amantadine, buspirone, L5hydroxy tryptophan, thyrotropinreleasing hormone, pregabalin, and several other medications, but this has not been confirmed in randomized studies. Cerebellar tremor was reported to be successfully treated with isoniazid and some anticonvulsants (clonazepam, carbamazepine, and topiramate). Stereotaxic thalamic surgery, including deep brain stimulation, may also be a good option in some cases. For instance, the mitochondrial 220 Part 7 Movement Disorders antioxidant idebenone was shown to reduce cardiac hypertrophy and, at higher doses, to improve neurological function, while ironchelating therapy with deferiprone and other agents has been shown to have some positive effect in pilot clinical studies and cell model experiments. It is directed at preventing various complications such as contractures or muscle atrophy, maximizing strength and conditioning, and improving coordination and gait. Special complexes of "cerebellar" or "sensory" exercises, as well as biofeedback with stabilography, are strongly recommended. A number of new molecular strategies for the treatment of different forms of hereditary ataxias are currently under development. Hopefully, in the future these technologies will offer significant advantages over any traditional therapy. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. Friedreich ataxia: Molecular mechanisms, redox considerations, and therapeutic opportunities. Although movement disorders related to antipsychotic medication were probably seen earlier, the causative effects of these drugs were not recognized until 1956, when the first report of chlorpromazineinduced movement disorder was made by Hall. While initially thought to be rare, it is now estimated to occur in up to 20% of patients exposed to these drugs. Clinical presentations may be acute, subacute, or delayed for months, and may mimic virtually every known hyper and hypokinetic movement disorder, either alone or in combination. This chapter focuses on several movement disorders caused by commonly prescribed drugs, excluding the neuroleptic malignant syndrome. Acute dystonia responds well to parenteral anticholinergics (biperiden, benztropine), antihistamines (diphenhydramine), or benzodiazepines (diazepam or lorazepam), although repeated doses may be necessary over a few days. Acute akathisia Acute akathisia (from the Greek meaning "inability to sit") is a very common and early doserelated side effect of neuroleptics and other dopamine receptor blocking drugs, although it is less frequently seen with atypical neuroleptics. Patients present with an aversion to keeping still and a subjective sensation of restlessness, accompanied by complex or stereotyped movements, including pacing, marching on the spot, picking at clothes, rocking, and crossing/uncrossing legs. The prevalence varies widely between studies, but acute akathisia is estimated to affect 40% of exposed patients, although this may be an underestimate. Half (50%) of affected individuals develop the condition within the first month, and 90% within 3 months of drug exposure. The pathophysiology of acute akathisia is not well understood, but it may result from a blockade of the mesocortical dopamine receptors. Treatment should comprise dose reduction, switching to a less potent drug, or, if possible, drug withdrawal, as this is the most effective treatment. If this is not possible or not effective, the addition of various drugs including anticholinergics, amantadine, propranalol, clonidine, benzodiazepines, and, more recently, opioid agonists (such as propoxyphene) has been reported to be effective. Traditionally, betaadranergic blockers such as propranolol have been used as firstline agents to treat akathisia. Anticholinergics are probably not as effective and are limited by doserelated anticholinergic side effects. Several placebocontrolled trials have demonstrated benefit from benzodiazepines, including clonazepam (0. Clinical manifestations vary with age: children often develop generalized spasms, sometimes with an axial emphasis. Dystonic spasms and postures tend to have an abrupt onset and are frequently painful and fluctuating in their distribution. Acute choreoathetosis A wide variety of commonly used drugs have been reported to cause choreoathetosis, raising the question of individual susceptibility as a consequence of prior basal ganglia damage.
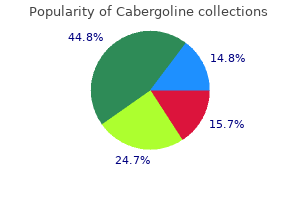
| Comparative prices of Cabergoline | ||
| # | Retailer | Average price |
| 1 | Hy-Vee | 771 |
| 2 | SonyStyle | 949 |
| 3 | Gap | 653 |
| 4 | Bon-Ton Stores | 856 |
| 5 | QVC | 375 |
| 6 | HSN | 352 |
Purchase generic cabergoline line
By blocking presynaptic dopamine reuptake and also by altering postsynaptic receptor sensitivity pregnancy massage cheapest cabergoline, cocaine may cause or trigger chorea and other hyperkinetic movements. Choreoathetosis and, less frequently, tremor, asterixis, and myoclonus have also been reported with phenytoin and anticonvulsants such as carbamazepine, felbamate, and gabapentin. Acute tics Druginduced tics are indistinguishable from those seen in Tourette syndrome and are caused by drugs that enhance dopaminergic transmission. Because tics are not always exacerbated or caused by central stimulants, undefined predisposing factors may also be important to this drug effect. Klawans and colleagues reported that patients may take 18 months or more to recover. In most cases, it is reversible once the offending drug is withdrawn, although as mentioned this may take 18 months or more. If withdrawal of the offending drug is not possible, it may be replaced by an atypical neuroleptic such as clozapine or quetiapine. If symptomatic treatment is necessary, levodopa may help in up to 30% of cases, although at the risk of aggravating the underlying psychiatric disorder. Some patients respond well to anticholinergics and amantadine, but these may worsen psychotic symptoms or result in confusion in elderly patients. Other options include dopamine agonists, propranolol, and electroconvulsive therapy. The risk is smaller for atypical neuroleptics, olanzapine and clozapine, the latter almost only occurring at doses above 250 mg. While it is said that some patients present symmetric akinetic rigid parkinsonian syndrome without tremor, these represent the minority. They have been reported most often in association with drugs that interfere with dopaminergic transmission, although seldom with drugs that act presynaptically, such as reserpine or tetrabenazine. Prevalence rates vary widely, probably reflecting the differences in patient samples and diagnostic criteria. There are several clinical subtypes of tardive dyskinesia, which may occur alone or in combination. The orobuccoliguomasticatory syndrome is the most common and first described subtype, representing 40% of patients in one study. It is characterized by stereotyped movements of tongue twisting and protrusion, along with facial grimacing. The clue to the diagnosis (apart from the history of drug exposure) is the not infrequent coexistence of other typical tardive movement syndromes, such as tardive tics, myoclonus, and tremor. Risk factors for developing tardive dyskinesias include chronic neuroleptic therapy (especially polypharmacy), age greater than 40, chronic schizophrenia, female sex, and the indiscriminate use of anticholinergic medication. On the other hand, the newer atypicals have a higher risk of producing metabolic syndrome. This leads to dopamine receptor supersensitivity, in which amounts of dopamine normally too small to induce dyskinesias are able to do so. Dopamine receptor blockers should be used for as short a time as possible and in the lowest dosage possible. Once tardive dyskinesia has developed, the offending drug should be withdrawn, although this is often not possible without relapse of the psychiatric illness. Improvement occurs in most cases, but complete and persistent resolution is seen in only 2%. The switch to atypical dopamine receptor blockers such as clozapine has been proposed as a desensitization technique and may be helpful in up to 40% of cases based on uncontrolled studies (controlled studies are lacking). Withdrawal of anticholinergics is recommended for orofacial dyskinesias, although they may help in the treatment of tardive dystonia. Further reading 57 Paroxysmal movement disorders Sopio Sopromadze1 and Alexander Tsiskaridze2 1 2 Department of Neurology, N. Kipshidze Central University Clinic, Tbilisi, Georgia Department of Neurology, Ivane Javakhishvili Tbilisi State University, Tbilisi, Georgia Paroxysmal movement disorders are a heterogeneous group of rare conditions characterized by episodic dyskinesias with sudden onset and brief duration. Paroxysmal dyskinesias (PxDs) can occur spontaneously or may be precipitated by sudden movements, prolonged exercise, caffeine and alcohol consumption, emotional stress, or fatigue. Abnormal movements can present with dystonic, choreic, ballistic, and other presentations, or a combination of these hyperkinesias. The current classification of PxDs by Demirkiran and Jankovic from 1995 is based on the precipitating events (Table 57. Initially hypnogenic paroxysmal dyskinesia, a fourth form of paroxysmal disorder, was included in the PxD classification. However, recently it has become clear that hypnogenic paroxysmal dyskinesia is a form of frontal lobe epilepsy in which dyskinesias occur only at night during sleep. Besides these three forms of PxDs, which are of genetic origin, occur without loss of consciousness, and are characterized by normal cerebral imaging, symptomatic PxDs have been described in patients with multiple sclerosis, stroke, trauma, encephalitis, metabolic disorders, cerebral neoplasms, and migraine. Later, in 1967, Kertesz described a new episodic disorder termed "paroxysmal kinesigenic choreoathetosis," in which episodes were very brief and induced by sudden movements. In 1977, Lance described a third form of attacks lasting between 5 and 30 minutes and provoked by prolonged exercise but not by sudden movements, which was further classified as "paroxysmal exerciseinduced dyskinesia. It is considered a familial disorder with autosomal dominant inheritance and incomplete penetrance, but sporadic cases often occur. Historical aspects the first description of a patient with PxD was made in 1940 by Mount and Reback and the term "paroxysmal choreoathetosis" appeared in the literature for the first time. Whether these conditions are of an epileptic origin or represent a basal ganglia disorder is as yet unknown. Concerning cellular mechanisms, channelopathy has been considered the main pathophysiological hypothesis for a rather long time. Indeed, paroxysmal dyskinesias have many clinical similarities to other episodic disorders of the nervous system, such as episodic ataxias and periodic paralysis. Both conditions have an early age of onset and there is a tendency for both to abate in adulthood. Recently genetic causes of various forms of paroxysmal dyskinesias have been elucidated. However, there is marked pleiotrophy of mutations in such genes, which expands the spectrum of clinical manifestations. It is also noteworthy that not all patients with a clinical picture of PxDs have mutations in these genes. It catalyzes the detoxification of methylglyoxal, which is reported to be neurotoxic. Clinical features PxDs represent a group of episodic abnormal involuntary movements manifested by recurrent attacks of dystonia, chorea, athetosis, or a combination of these disorders. The movements can be any combination of chorea, athetosis, ballism, and/or a dystonic posture. Some patients manage to suppress the attack during the aura by holding the affected limb tightly, by moving slowly, or by crossing the legs. Family history is present in 27% of cases and usually follows an autosomal dominant pattern of inheritance. Overall prognosis for the idiopathic form is good and the disease often abates in adulthood, as the attack frequency decreases with age. Patients often have a combination of involuntary dystonic, choreatic, athetotic, and ballistic movements, mainly affecting the limbs, often unilateral or asymmetric. Involuntary movements occur after continuous exercise such as walking, swimming, or running. Dystonic movement affecting lower limbs, often bilaterally, is the most common feature.
Discount cabergoline 0.5 mg with mastercard
Restricted women's health center birmingham al buy genuine cabergoline online, non productive infection of astrocytes contributes to neuronal dysfunction. Clinical features Asymptomatic neurocognitive impairment is, by definition, an asymptomatic condition with a normal clinical neurological examination. The diagnosis requires formal neuropsychological testing revealing decreased performance in two or more domains. Cognitive domains initially affected include verbal and visual memory (retrieval rather than recognition), complex sequencing, mental flexibility, and visual construction. Patients with mild neurocognitive disorder may complain of mild neurocognitive impairment and minor functional impairment in daily living. These patients usually are unable to complete complex tasks both at work and at home, but may continue to work, albeit at a decreased level of efficiency. This presents clinically with impaired shortterm memory, poor concentration and attention, and executive dysfunction with mental slowing and impaired judgment. Mild early motor symptoms become noticeable and consist of psychomotor slowing, leading to difficulties with fine finger movements and balance problems. Subtle gait difficulties are present at this stage, resembling impaired postural reflexes as seen in patients with extrapyramidal disease. At this stage of illness, neurological examination is normal except for mild slowing of repetitive movements. Behavioral abnormalities include apathy, inertia, loss of libido, irritability, blunting of emotional responses, and waning interest in work and hobbies, ultimately leading to social withdrawal. As disease progresses, spasticity (especially of lower limbs), clonus, frontal release signs, tremor, and ataxia develop. In advanced dementia, signs of cooccurring myelopathy and/or peripheral neuropathy may contribute to abnormal motor findings. Focal neurological signs such as hemiplegia, hemianopia, and hemisensory impairment are absent; in fact, their presence suggests other pathologies. In late or endstage disease, dementia becomes global, with mutism, abulia, and incontinence. Investigations Screening tests Neuropsychological testing can be used for screening purposes in highrisk asymptomatic or early symptomatic patients and for 336 Part 9 Prion Diseases and Neurovirology followup evaluation in patients with established cognitive impairment. Appropriate normative standards are not available for large parts of the developing world. The revised cutoff score of 14 out of a possible 16 points is sufficiently sensitive and specific. Age inappropriate cerebral atrophy with corresponding ventricular enlargement is a typical finding. Increasing ventricular size consistent with subcortical tissue loss mirrors progressive clinical deterioration. T2 weighted image showing confluent whitematter hyperintensities, sparing the subcortical U fibers. Reproduced with permission of the American Association for the Advancement of Science. These and other studies showed that clinical myelopathy is uncommon, even though frequently described at autopsy. Oxidative damage to oligodendrocyte membranes causes increased consumption of antioxidants. Motor features include symmetric lower limb weakness with hyperreflexia, spasticity, and extensor plantar responses. Sensory findings are less prominent, although loss of vibration and position sense result in sensory ataxia. This combination can lead to a mixed clinical picture where lower limb hyperreflexia and extensor plantar responses are seen in conjunction with absent ankle jerks. Somatosensory evoked potentials may demonstrate prolonged central conduction time, indicative of spinal cord involvement. The effect of highdose supplemental methionine was evaluated in a placebocontrolled phase 2 trial on 56 patients. There was a modest, but not significant improvement in central conduction time, but no benefit clinically. Symptomatic treatment of spasticity, urinary urgency, and erectile dysfunction is warranted. These disorders may result from the virus itself, through indirect, immunemediated mechanisms, or may be the result of antiretroviral medications. Additionally, opportunistic infections can produce neuropathic and myopathic symptoms. A crosssectional study of eight Asia Pacific countries found a comparatively lower prevalence of neuropathy at a rate of about 20%. Risk factors identified in these studies also include greater height and age, as well as exposure to stavudine. Examination reveals diminished sensation in a stockingglove pattern, reduced or absent ankle jerks, and sensory ataxia. Patients report difficulty rising from a seated position or trouble climbing stairs. Examination reveals weakness, greater proximally, typically with preservation of sensation and normal reflexes unless there is concomitant neuropathy. If there is an opportunistic infection underlying the myopathy, such as toxoplasmosis, then there may be additional clinical signs and symptoms resulting from a focal central nervous system lesion. To determine whether symptoms are medication induced, it is necessary to stop the potentially offending drug and observe the patient for improvement; however, it may take months to see any change. Furthermore, symptoms may worsen before they improve, a phenomenon known as "coasting. Electrodiagnostic studies may be normal if predominantly small fibers are affected, and in that case skin biopsy may aid in diagnosis by showing a reduction in the intraepidermal nerve fiber density. Among symptomatic treatments, only the 8% transdermal capsaicin patch and an alternative treatment, smoked cannabis, have proven efficacy. These include topical lidocaine, gabapentin, lamotrigine, pregabalin, amitriptyline, duloxetine, peptide T, and mexiletine. Neuroregenerative treatments, such as acetyl Lcarnitine, prosaptide, and recombinant human nerve growth factor, have been tested without positive results or clinical applicability. Electrophysiology may yield evidence of demyelination with slowed conduction velocities, conduction block, or prolonged distal latencies; there may also be absent or prolonged late responses. Patients present with progressive weakness, sensory loss, pain, and urinary dysfunction resembling a cauda equina syndrome, including the typical saddle distribution of pain and sensory loss. The virus may infect the cauda equina leading to inflammation of the lumbosacral nerve roots; in some cases the arms and cranial nerves may be affected. Despite treatment with ganciclovir, often combined with foscarnet, the prognosis is poor. Mononeuropathy multiplex the presentation of mononeuritis multiplex is variable, with occurrence of multiple mononeuropathies, typically described as painful "stepwise" deficits. For example, up to onethird of patients with cryptococcal meningitis in developing countries also have tuberculous meningitis. The best clinical approach to these conditions rests on neurological evaluation to decide whether the disease is diffuse or focal. This chapter focuses on infections that are common and some of these infections, such as varicella zoster virus and neurosyphilis, are discussed in further detail in this textbook and in other more specialized texts. These conditions differ in terms of epidemiology, pathophysiology, clinical features, and treatment, and are therefore discussed separately. Transmission is primarily via the respiratory route, with deposition of yeast spores into the pulmonary alveoli followed by phagocytosis by alveolar macrophages. Cryptococcal neoformans pulmonary disease can be asymptomatic and therefore meningitis may be the presenting feature.
Buy cabergoline with mastercard
Human rabies immunoglobulin (Ig) is administered to patients without prior vaccination up to 7 days post exposure; given after this point women health issues discount cabergoline master card, Ig administration could suppress antibody production from vaccination and is therefore not recommended. Supportive treatments include benzodiazepines, ketamine, barbiturates, and morphine. Nimodipine to relieve vasospasm should be used with caution, since it may cause hypotension and shock. Pathophysiology After the bite of an infected mosquito, the virus multiplies in skin and lymph nodes, followed by transient viremia. In the brain, the virus causes diffuse encephalitis, seen macroscopically in autopsy specimen as focal petechiae or hemorrhage involving gray matter. Microscopically, there is perivascular cuffing, mononuclear cell infiltration, and phagocytosis of dead cells. Immunologically, the patient mounts a rapid and effective IgM response within days, which limits viral replication and may facilitate lysis of infected cells. Clinical features Only 1 in 25 to 1 in 1000 patients infected with Japanese encephalitis virus becomes symptomatic. Symptoms range from mild, nonspecific, flulike illness to fatal meningoencephalitis. Severity depends on the route of viral entry, the size and virulence of the inoculum, and host factors such as age, prior immunity, genetic factors, and general health. Titubation, pillrolling tremor, choreoathetosis, orofacial dyskinesia, and upper motor facial nerve palsy are seen in about 10% of patients, presumably due to the involvement of the thalamus and basal ganglia. Opisthotonus, opsoclonus, myoclonus, changes in respiratory pattern, decerebrate or decorticate posturing, and pupillary and vestibuloocular reflex abnormalities indicate brainstem involvement and may herald a poorer prognosis. Some patients present with flaccid paralysis of one or more limbs, probably due to anterior horn cell damage, and onethird of these eventually develop encephalitis. Singlephoton emission tomography shows hyperperfusion in the thalamus and putamen. Previous infection by or vaccination against another flavivirus could cause crossreactivity. Japanese encephalitis Epidemiology Japanese encephalitis virus, a flavivirus, is transmitted by the Culex tritaeniorhynchus mosquito between birds and pigs, both of which develop high viremia and are important in maintaining, amplifying, and transmitting the disease. Humans do not develop significant viremia and are thus deadend hosts, but can become infected when living close to the enzootic cycle of the virus in rural areas. Japanese encephalitis has spread from East and Southeast Asia to Russia, the Guam islands, India, and the northern tip of Australia. Today, it is the most common cause of arboviral encephalitis, with an annual incidence of 1. It causes some 67,900 infections and at least 15,000 deaths annually, accounting for more infections and death than all other arboviruses combined. In northern Asia, epidemics occur during the summer months; in the south, the disease is endemic, with outbreaks occurring in the monsoon months. Up to 20% of vaccine recipients report local side effects, such as swelling, tenderness, and redness, while 10% report mild systemic side effects of fever, chills, malaise, and headache. The risk of severe neurological side effects, such as encephalitis or encephalopathy, seizure, and peripheral neuropathy, is only 0. Recently, up to 1/100 recipients in Europe, North America, and Australia reported a new pattern of side effects consisting of pruritus, urticaria, and facial angioedema. Production of this vaccine has ceased and the remaining stock in the United States is reserved for children. Although it has been shown to be effective and safe in children, it is only available in some Asian countries. Travelers who visit endemic areas during rainy seasons, or who stay near pig farms, or in rural or agricultural areas or for long durations (30 days), need to be vaccinated. There is no definitive antiviral agent for Japanese encephalitis, although various compounds, such as isoquinoline and interferon, have been tried. Histologically, microglial nodules with neuronal loss are seen in the gray and white matter of the cerebrum, hippocampus, thalamus, medulla, and, possibly, anterior horn cells. There is perivascular infiltration by mononuclear cells and focal inflammation of cranial nerves and leptomeninges. Immunohistochemical staining shows viral antigens in neuronal cytoplasm and processes associated with glial nodules. Clinical features In endemic areas, over 80% of infection is asymptomatic, and most symptomatic infections are mild, selflimiting febrile illnesses. Half of patients have a nonpruritic roseolar or maculopapular rash that generally lasts no more than a week. In these patients, 30% develop meningitis while 60%, particularly those 50 years of age, develop encephalitis. In those with encephalitis, common symptoms include fever, headache, nausea, vomiting, chills and rigors, myalgia, backache, and weakness. Common signs are confusion, drowsiness, tremor, neck stiffness, myoclonus, ataxia, parkinsonism such as rigidity, bradykinesia, and postural instability, cranial nerve palsies such as dysphagia and absent corneal and gag reflexes, and brainstem dysfunction such as nystagmus and apnoeic episodes. Some patients develop an asymmetric flaccid paralysis with areflexia and bladder dysfunction due to anterior horn cell involvement. Investigations In patients with encephalitis or meningitis, mild peripheral blood leukocytosis is common. Electromyography may show axonal neuropathy, probably reflecting anterior horn cell involvement, although a demyelinating neuropathy is also seen in some patients. Because West Nile virus crossreacts with antibodies directed against other flaviviruses, the presence of specific IgM should be confirmed via plaque reduction assay to detect neutralizing antibodies. West Nile encephalitis Epidemiology Birds are natural reservoirs of West Nile virus, a flavivirus transmitted by the Culex mosquito from birds to humans and animals such as horses, domestic animals, and other mammals, which are deadend hosts. The virus is widely distributed across Africa, the Middle East, India, Southern Europe, Russia, and North America. Although it is asymptomatic in endemic populations, outbreaks have occurred in South Africa, Europe, and Russia. Phylogenetic studies reveal at least five lineages of the virus, with lineage 1 clade 1a found in North America, Europe, the Middle East, and Africa, generally causing more severe human neurological disease and being responsible for almost all recent outbreaks. Lineage 2 (endemic in subSaharan Africa and Madagascar, with occasional outbreaks in Europe) and lineage 1 clade 1b (also known as Kunjin virus, found in Australia) generally cause milder disease. Lineage 3 contains of a single isolate from Chapter 80 Acute viral encephalitis 311 Treatment Apart from supportive management, there is no specific treatment for West Nile encephalitis.
Purchase cheap cabergoline line
Immunoglobulin replacement can be of value in addition to measures directed towards the primary abnormality breast cancer 7 cm buy line cabergoline. IgG subclass deficiency, the heavy chain genes are intact, indicating that the lack of subclass is due to other genetic factors controlling transcription or translation. Where the patient is symptomatic with recurrent infections and evidence for poor or absent immunization responses, immunoglobulin replacement is effective in preventing infection. Selective IgA deficiency IgA deficiency is the most common form of immunodeficiency among Caucasians, occurring in about one in 600 of the population. Most cases are sporadic but some have family members with varied forms of antibody deficiency. It has been noted in patients treated with phenytoin or penicillamine, although the underlying susceptibility to develop IgA deficiency may be part of the primary disease for which they receive these treatments, for example, epilepsy or rheumatoid arthritis. IgA deficiency is associated with an increased incidence of autoimmune diseases such as juvenile arthritis, systemic lupus erythematosus, coeliac disease and pernicious anaemia. Almost all IgAdeficient patients possess circulating B cells bearing surface IgA, but these appear immature, often coexpress IgM and fail to differentiate into IgAsecreting plasma cells. In some cases, plasma cells producing the IgA2 subclass are present in the gut with the defect confined to IgA1producing bone marrow plasma cells; in others, both subclasses are deficient. Circulating T cells, which block the differentiation of IgA plasma cells, have been identified in some patients with IgA deficiency. The most common IgG subclass abnormality in adults is deficiency of IgG3, whereas in children it is IgG2. Serious infections may be associated with IgG2 deficiency even when the total serum IgG level is within the normal range, although some individuals with IgG2 deficiency due to deletion of the 2 gene are entirely healthy. Recurrent bacterial infection and asthma may complicate IgG3 deficiency, but the mechanism is poorly understood. In most cases of Specific antibody deficiency with normal immunoglobulins There are welldescribed patients who have normal levels of serum immunoglobulins and IgG subclasses but still suffer from recurrent infections typical of antibody deficiency. These patients may be shown to Chapter 11 Primary and secondary immunodeficiency disorders 137 be immunologically unresponsive to bacterial polysaccharide antigens such as pneumococcal capsular polysaccharide. This can be demonstrated by measuring the level of antibodies before and after immunization with pneumococcal polysaccharide vaccine (Pneumovax). Such patients lack isohaemagglutinins and are particularly susceptible to meningitis and septicaemia with encapsulated organisms, for example, pneumococci and meningococci. Defects of mobility and phagocytosis Various abnormalities have been described which affect the ability of phagocytes to migrate to sites of infection. Immune adherence is intimately linked to phagocytosis and it is rare to find defects of phagocytosis alone. In the last case, neither IgA nor secretory component is detectable in saliva or jejunal fluid, whereas the serum IgA level is normal. Phagocyte defects Phagocytes, that is, neutrophil polymorphs and monocytes, have a critical role in defence against many bacterial pathogens. Phagocyte function divides into three sequential components: (i) mobility, margination and adherence, (ii) phagocytosis and (iii) intracellular killing (see Chapter 7). The primary phagocyte defects described in the following illustrate the importance of these different processes (Table 11. It is important to realize, leucocyte adhesion deficiency these patients usually present with infections of the skin, mouth, respiratory tract and around the rectum but with little evidence of pus formation. Wound healing is impaired and delayed separation of the umbilical cord is a feature of most cases. This condition is characterized by a failure of neutrophils and monocytes to migrate to sites of tissue infection (in spite of persisting neutrophilia). The condition is usually inherited as an autosomal recessive and several different mutations Table 11. They contain giant secondary lysosomes which contain the products of fusion of many cytoplasmic granules, and the functional abnormalities observed may well be a consequence of the continuous activation which these cells undergo. Cyclosporin may be helpful, and bone marrow transplantation and intravenous immunoglobulin have been used successfully. The effect of infection has already been emphasized, but impairment also occurs in malnutrition, burns, diabetes and uraemia and following the administration of various drugs and anaesthetic agents. Phagocytosis is normal but oxidative killing is absent in both neutrophils and monocytes. The molecular cause of most cases of membrane oxidase deficiency is defective production of the heavy chain of the cytochrome b involved in the electron transport chain linked to the membrane oxidase. The dermatitis and proneness to infection are usually evident within the first 6 weeks of life. Stem cell transplantation, preferably early in childhood, using either a matched sibling donor or a matched unrelated donor is curative. In the former case, the enzyme is deficient in neutrophils and monocytes although eosinophil peroxidase is preserved. This shows a draining fungal abscess on the chest wall (Scedosporium apiospermum) in a patient with chronic granulomatous disease. Complement deficiencies Heritable deficiencies of each of the nine components of the classical pathway (including the three subunits of C1) as well as properdin, factor D and the inhibitors of C1 (C1 esterase inhibitor) and C3 (H and I) have been identified in man (see Clinical Case Scenario 6. Major deficiencies of C3 are associated with severe bacterial infection; deficiencies of components of the membrane attack pathway give increased susceptibility to infection with Neisseria, for example, gonococci and meningococci, whereas deficiencies of early components in the classical pathway are more commonly associated with immune complex disease rather than overt infection (Table 11. Genetic abnormalities of factor H and factor I have been associated with the development of the haemolytic uraemic syndrome. This protein binds to mannose and Nacetylglucosamine on microbial cell walls and activates C3 by a C1qindependent route (see Chapter 6). In contrast, organisms which generate H2O2 and are catalase negative, for example, H. The reason for this disparity is that hydrogen peroxide produced in the absence of catalase can be incorporated into the metabolic pathway of the phagocyte to compensate for the lack of endogenous H2O2 production so that, in effect, the pathogen commits suicide (Table 11. The cleavage products of C3 are preeminent in the defence against pyogenic bacteria, and the fact that only major deficiencies of C3 associate with severe infection emphasizes the importance of the alternative pathway in achieving C3 conversion when classical pathway components are deficient (see Chapter 6). The association of defects in the classical pathway with immune complex disease is a consequence of the role of these components in inhibiting the precipitation of immune complexes and facilitating their clearance from the circulation (see pp. Oedema of the gut can present as severe abdominal pain, and laryngeal oedema can prove fatal. The large majority of patients are heterozygous for C1 esterase inhibitor deficiency, but their inhibitor levels are usually well below 50% of normal because of increased catabolism. The condition can be treated by giving inhibitors of fibrinolysis, for example, tranexamic acid, or the administration of attenuated androgenic steroids such as danazol and stanozolol. Purified or recombinant C1 esterase inhibitor is now available for replacement therapy by intravenous injection, and recently, a bradykinin receptor blocker, icatibant, has been shown to be very effective in treating acute attacks. This has been variously attributed to absorption of the inhibitor protein by tumour cells, the formation of antiidiotype complexes or the presence of a monoclonal autoantibody, which inactivates the inhibitor. The most common cause of angiooedema in the older population is due to the use of the anti hypertensive drugs belonging to the class of angiotensin converting enzyme inhibitors, which prevent the breakdown of bradykinin. Chapter 11 Primary and secondary immunodeficiency disorders 141 Secondary forms of immunodeficiency the most common cause of serious immunodeficiency in clinical practice is none of the aforementioned but rather the impact that a number of diseases and the therapies used to treat them have upon the immune system. Other disorders associated with impaired protein intake or protein loss also cause immunodeficiency, and secondary impairment develops during the course of most forms of chronic inflammatory disease. Removal of the spleen, however, greatly increases the risk of fulminant infection with pneumococci, meningococci, Table 11. Intraerythrocytic pathogens, which would normally be phagocytosed in the spleen, such as Plasmodium (causing malaria), Bartonella and Babesia also cause severe disease. Polyvalent pneumococcal, Hib and meningococcal vaccines should be given (and, where possible, before splenectomy is performed) in conjunction with lifelong prophylactic antibiotics, usually penicillin V or erythromycin. The immunological changes consist of impaired clearance of intravascular organisms, reduced concentrations of serum complement and IgM, and a disturbance of the normal profile of lymphocyte subpopulations, often with a moderate lymphocytosis. The immune system has a limited capacity for extra thymic Tcell development and considerable capacity for extrathymic Tcell expansion.
0.25mg cabergoline
Localization the tumours have all occurred in the larynx menstruation 9 dage discount cabergoline 0.5mg with visa, with no site of predilection 744. Clinical features Progressive dysphonia and dyspnoea are the most common complaints. Histopathology the hallmark is the presence of numerous, non-cohesive, bizarre giant cells that contain prominent, frequently multiple nuclei with coarse chromatin and large nucleoli. The cytoplasm is abundant, eosinophilic, sometimes vacuolated, and often contains neutrophils or cellular debris. Additionally, the tumour contains a background population of smaller anaplastic tumour cells. Clusters of pleomorphic giant cells in a background with admixed foamy histiocytes and inflammatory cells. Prognosis and predictive factors the reported cases have shown a poor prognosis 744. Eveson In a large series of laryngeal tumours from one institution, 72% of salivary gland-type neoplasms were malignant 1039. They are much more common in men and most cases present between the ages of 45 and 75 years (peak incidence in the 6th decade) but cases have been reported in children 1750. Nearly half of the cases present with, or develop, cervical lymph node metastases 217,533. The majority are subglottic (60%) or supraglottic (35%) and the true cords are involved in only 6% of cases 1573. Tumour types No of cases 3 12 1 11 11 19 57 % 5 21 2 19 19 33 Pleomorphic adenoma Oncocytic tumours Cystadenoma Mucoepidermoid carcinoma Adenoid cystic carcinoma Adenosquamous carcinoma Total *Modified from Heffner 1039 Other salivary-type tumours A variety of other carcinomas have been reported in the seromucous glands of the larynx. However, they are very rare and are usually presented as single case reports: acinic cell carcinoma 734,1250, 2146,2454, malignant myoepithelioma 1167, carcinoma ex pleomorphic adenoma 180,232,1729,2220, epithelial myoepithelial carcinoma 1726, salivary duct carcinoma 745,910, papillary adenocarcinoma 2182, mucinous adenocarcinoma 2640 and clear cell carcinoma 1855,2020,2306. Barnes Neuroendocrine neoplasms of the larynx are a heterogeneous group of tumours that vary from benign to highly malignant. As a group, they are uncommon with only about 500 cases recorded in the literature as of 1998 738. The atypical carcinoid is the most frequent, constituting 54% of all neuroendocrine tumours in this site, followed by the small cell carcinoma, neuroendocrine type (34%), paraganglioma (9%) and the typical carcinoid (3%) 125,648,897, 2420,2816. Cells similar if not identical with Kultchitsky cells of the bronchi are found in the larynx. These, as well as pluripotential endobronchial stem cells, are the putative cells of origin of the typical carcinoid, atypical carcinoid, and small cell carcinoma, neuroendocrine type. Paragangliomas of the larynx are derived from paraganglia normally found in the larynx and are discussed in chapter 8. Combined small cell carcinoma, neuroendocrine type, with non-small cell carcinoma (squamous cell carcinoma, adenocarcinoma,etc. Paraganglioma Non-chromaffin paraganglioma atypical carcinomas may fulfill the diagnostic criteria of large cell neuroendocrine carcinoma of lung 2Not all small cell carcinomas of the larynx will show neuroendocrine differentiation 1Some Typical carcinoid Definition An epithelial tumour of low-grade malignancy composed of round to spindle cells with histologic, immunohistochemical and ultrastructural evidence of neuroendocrine differentiation. It is three times more common in men and most patients are between 45-80 years of age (average 64 years) at diagnosis 738, 2419. Typical carcinoid of epiglottis composed of small trabeculae and clusters of cells lying in the lamina propria. Localization Most occur in the supraglottic larynx in the vicinity of the aryepiglottic fold, arytenoid or false vocal cord. Clinical features Symptoms, ranging from three weeks to four years in duration, include dysphagia, hoarseness and a sore throat. At least one patient developed the carcinoid syndrome after the tumour metastasized to the liver 738. The distinction depends on the presence (oncocytic) or absence (oncocytoid) of mitochondria on ultrastructural examination. Prognosis and Predictive factors Since radiation and chemotherapy are ineffective, surgery is the treatment of choice. The extent of resection should be as conservative as possible, as long as complete removal is achieved. At least one patient developed the carcinoid syndrome and another died of disease five years after treatment. They are also variably positive for a variety of peptides, including serotonin, calcitonin, bombesin and somatostatin. Atypical carcinoid Definition An epithelial tumour composed of round to spindle cells with histologic, immunohistochemical and ultrastructural evidence of neuroendocrine differentiation exhibiting more mitoses and cellular atypia than a typical carcinoid. Cysts with intracystic papillary-like projections of tumour cells may also be seen. Some tumours may even fulfill the diagnostic criteria of large cell neuroendocrine carcinoma of the lung (10 or more mitoses per 10 high power fields and prominent necrosis). Mucinous changes, amyloid, spindle cells and oncocytic-oncocytoid cells may also be observed. It is 2-3 times more common in men and has been described in patients from 36-83 years of age (average 61 years) 2769,2816. Localization Over 90% arise in the supraglottic larynx in the vicinity of the aryepiglottic fold, arytenoid or false vocal cord. Clinical features Hoarseness, dysphagia, pain in the throat and a neck mass are the usual symptoms. Electron microscopy Membrane-bound, electron-dense neurosecretory granules ranging from 70420 nm are prominent 2769. Cellular junctional complexes, rough endoplasmic reticulum, mitochondria, Golgi complexes and infrequent bundles of tonofilaments may also be seen. Clinical and imaging studies to detect the presence or absence of a mass in the larynx or thyroid may offer some assistance. Reliance on this test to distinguish between these two tumours is, therefore, not absolute. Neuroendocrine neoplasms 137 Factors adversely affecting prognosis include metastatic disease at presentation, positive tumour margins, angiolymphatic invasion, and tumours larger than one centimeter 2769,2816. Small cell carcinoma, neuroendocrine type Definition A highly malignant epithelial tumour composed of small round, oval or spindle cells with evidence of neuroendocrine differentiation. The tumour (arrows) is ulcerated and indistinguishable from a squamous cell carcinoma From L. High magnification showing small spindle cells with hyperchromatic nuclei devoid of nucleoli. Clinical features Symptoms and signs are those associated with other laryngeal neoplasms and depend on the site of origin. Macroscopy the tumours often present as ulcerated submucosal lesions and, as a consequence, may be indistinguishable from ordinary squamous cell carcinoma. Metastases to cervical lymph nodes have observed in 43% of patients, to skin and subcutaneous tissues in 22%, and to other distant sites in 44% (particularly lungs, liver, and bones 2816. Surgery is preferred and, depending on the site of the tumour, requires a partial or total laryngectomy. Because of the high incidence of cervical lymph node metastasis, a neck dissection, even when the lymph nodes are clinically negative, is warranted 752. It is three times more common in men and is distinctly unusual in patients below 40 years of age 897. Malignant lymphomas are positive for leukocyte common antigen and negative for neuroendocrine markers. Almost half of patients will present with positive cervical lymph nodes and about 60-90% will develop distant metastases, especially to the lungs, liver and bones. Because many patients have disseminated disease at the time of diagnosis, radical surgery (laryngectomy with neck dissection) is rarely indicated. Most, if not all, have been associated with a squamous cell carcinoma, either in-situ or invasive 1201. The altered epithelium shows a variety of cytological and architectural changes that have traditionally been grouped under the term dysplasia. Rarely, malignant transformation can develop even from morphologically normal epithelium. Atypia has been used in the context of inflammatory and regenerative changes particularly referring to cytologic features. In this text, the term atypia refers to cytological change that may or may not be pre-malignant. Various classifications have evolved to describe the spectrum of histological changes in relation to their malignant potential 222,504,846,1054,1055,1253, 1254,1711,2317. Epidemiology the entire spectrum of laryngeal and hypopharyngeal precursor lesions are mostly seen in the adult population and affect men more often than women.
