

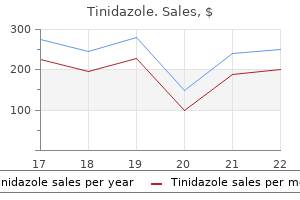
Order 1000mg tinidazole
Contusions are the result of a direct injury and involve the skin and the subcutaneous tissues as well as underlying muscle antibiotics for acne doxycycline discount tinidazole express. It is always important to order radiographs of the pelvis rather than individual views of the right or left hip. There is usually a history of trauma, and the location is readily apparent because of soft tissue swelling, ecchymoses, and pain. Most of these injuries occur during athletic activities, but they can also be the result of falls or other minor injuries. In the absence of an associated physeal injury or other fracture, treatment is typically immobilization with a gradual return to activity. In sprains, the physical examination typically reveals that the involved ligament is tender to direct palpation. Strains involve the muscles, and there is usually tenderness to palpation, soft tissue swelling, and pain with joint motion as a result of stretching of the involved muscle. A palpable defect within the muscle is uncommon except in the most severe injuries. These injuries usually limit the excursion of the muscle and its associated joints. In children who sustain sprains, strains, or significant contusions, anteroposterior and lateral radiographs should be obtained. Therefore, a physeal injury may be present and may have the same clinical features as a sprain. This condition should be suspected when there is more tenderness to palpation over the lateral malleolus than over the ligaments. If plain radiographs are normal, stress radiographs may be necessary to establish the diagnosis. Occult fractures of the tibia are a relatively common cause of limping or refusal to bear weight in very young children. These fractures can be the result of very innocuous trauma, such as tripping while walking, stepping on a toy, or falling from a height. Frequently, the injury may not have been observed, and the child cannot convey to the parents what happened, confounding diagnosis. This is an oblique fracture of the distal third of the tibia without an associated fibula fracture. Occult tibia fractures can also occur in the metaphyseal regions, usually distally, but only rarely in the diaphysis. There is mild tenderness and perhaps increased warmth on palpation over the fracture. If plain radiographs are normal, the child has no systemic symptoms, and an occult fracture of the tibia is suspected, simple immobilization in a long-leg cast is indicated. Another set of radiographs in 1-2 weeks usually reveals the fracture and evidence of healing. Benign and malignant neoplastic lesions that involve bone, cartilage, or soft tissue of the spine, pelvis, and lower extremities can manifest as a mass, can cause pain, and can produce an antalgic gait. Leukemia or metastatic neuroblastoma of the bone marrow may produce deep bone pain and limp without objective findings of swelling or tenderness on physical examination. Night pain is a common characteristic of both benign and malignant primary or metastatic tumors. The most common benign lesions that produce limping include a unicameral (simple) bone cyst and osteoid osteoma (Table 34. Other less common benign lesions that can produce pain and limping include eosinophilic granuloma of the bone, osteochondroma, and chondroblastoma. Chondroblastoma typically involves the epiphysis, especially of the proximal humerus. In unicameral bone cysts, the symptoms are usually caused by a nondisplaced pathologic fracture. The most common location for a unicameral bone cyst is the proximal humerus, followed by the proximal femur. Osteoid osteomas have a highly vascularized nidus, which incites an intense, painful, inflammatory reaction that produces sclerosis of the surrounding bone. Most benign neoplasms are visible on anteroposterior and lateral radiographs of the symptomatic area. Characteristics of benign lesions include well-circumscribed lesions without periosteal new bone formation or soft tissue mass. If a lesion is suspected but not visible on plain radiographs, such as may occur in an osteoid osteoma, a technetium bone scan may be helpful. Diagnosis is further aided by surgical biopsy, which may also allow for surgical treatment. Leukemia is the most common childhood malignancy and is frequently accompanied by musculoskeletal complaints, such as limping, fever, bone pain, pallor, bruising, and weight loss. Common malignancies involving the musculoskeletal system include osteogenic sarcoma, Ewing sarcoma, and intraspinal tumors, such as astrocytomas (Table 34. Intraspinal tumors tend to produce neurologic symptoms, such as muscle weakness, as the cause of limping. The other lesions may produce a mass, bone weakness, and possible pathologic fractures. A careful musculoskeletal and neurologic examination is necessary for any child with a suspected neoplasm. In many cases, a mass, either in the involved bone or in adjacent soft tissues, may be palpable. These lesions are frequently adjacent to joints and may result in decreased range of motion. Neurologic evaluation may show evidence of muscle weakness or abnormal reflexes, suggestive of spinal cord or peripheral nerve involvement. Anteroposterior and lateral radiographs of the involved area usually reveal the presence of a neoplasm. The preoperative diagnosis was Ewing sarcoma, but at biopsy the diagnosis was acute lymphoblastic leukemia. Radiographic abnormalities associated with acute leukemia include diffuse osteopenia, metaphyseal bands, periosteal new bone formation, geographic lytic lesions, sclerosis, and permeative distraction. When the infection is confined to the synovium of a joint, the condition is termed septic arthritis. If the primary focus of the infection is within bone, even if the joint is secondarily involved, the condition is termed osteomyelitis. Bacterial pathogens are the most frequent cause of osteoarticular infections in children, with Staphylococcus aureus being the most frequent etiology. Beyond the neonatal period, Kingella kingae is the second most frequent cause in children under 5 years of age. In older children and adolescents with puncture wounds of the foot, Pseudomonas aeruginosa, S. Sexually active adolescents may develop septic arthritis as a result of gonococcal infections. Patients with sickle cell anemia may develop osteomyelitis as a result of Salmonella species or pneumococcal infection. Acute hematogenous osteomyelitis most commonly involves the femoral neck, the distal femoral metaphysis, and the proximal tibial metaphysis. Children with these infections may be acutely ill; many may just have fever, limp, and localized pain. Subacute osteomyelitis, which has very distinct manifestations, occurs most commonly in the knee. Radiographs show sclerotic metaphyseal lesions that occasionally cross the growth plate into the epiphysis. Children with acute bone and joint infections may exhibit the clinical signs of bacteremia and infection, including elevations in temperature, white blood cell count, erythrocyte sedimentation rate, and C-reactive protein level. When the hip joint is involved, the child holds the hip in a position of flexion, abduction, and external rotation.
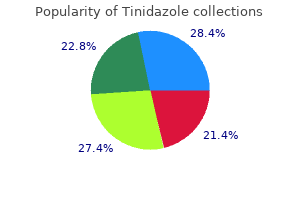
Order tinidazole 300 mg on-line
Anorectal manometry is a valuable diagnostic procedure if radiographic procedures are unrevealing antibiotics that treat strep throat order tinidazole 300mg otc. Normal internal anal sphincter relaxation with transient rectal distention rules out Hirschsprung disease. Paradoxical contraction of the internal anal sphincter suggests an absence of ganglion cells and is most common in Hirschsprung disease. Absence of relaxation has been noted in premature infants, in neonates with infection or sepsis, and in thyroid aplasia; normal function is seen after appropriate therapy. The sensitivity and specificity of this test varies somewhat among the different age groups (children versus infants versus neonates). A full-thickness rectal biopsy procedure is reserved for infants with bowel obstruction and for older children with abnormal rectal motilities and inconclusive suction biopsies. Appropriate therapy for specific lesion, then constipation management as needed No Yes Labs Normal Manometry Abnormal Abnormal Normal Constipation management Normal Constipation management Paradoxical internal anal sphincter contraction Rectal suction biopsy Hypothyroidism Hypercalcemia Lead intoxication Diabetes Infectious Hypopituitarism, etc. Appropriate therapy for underlying condition, then constipation management as needed Ganglion cells present Constipation management Transitional zone identified Full-thickness rectal biopsy at time of surgery for Hirschsprung disease Ganglion cells absent Liquid contrast enema Normal Diagnosis is based on the clinical manifestations in the absence of identifiable anatomic obstruction, as well as on motility studies that quantify abnormal bowel transit. Full-thickness intestinal biopsy should be reserved for severe or refractory cases, and care should be taken to pursue biopsy only if the patient is undergoing another indicated intraabdominal procedure, as biopsy in these patients increases the risk of postoperative adhesions and acquired obstruction. In anterior ectopic anus, the anal canal and the internal anal sphincter are displaced anteriorly in the perineum as a unit and are separated from the external anal sphincter, which remains posterior in its usual position. On physical examination, it may be possible to elicit an external sphincter anal wink in the usual location, posterior to the opening of the anal canal. In anterior anal displacement, the entire normal anal unit is located in the anterior perineum. Symptoms of constipation often begin in the neonatal period and are related to the difficulty in expelling stool through a canal that is angled anteriorly. If the displacement is severe enough to cause symptoms, surgical correction may be necessary to relocate the anus and relieve the obstruction. Failure to thrive examination Abdominal distention Poor growth Anal tone Rectal examination Malnutrition Laboratory Barium enema Uncommon Variable Rare Patulous Stool in ampulla Absent Massive amounts of stool, no transition zone Normal Anal Stenosis the diagnosis of anal stenosis may be delayed beyond the neonatal period, especially if the degree of stenosis is not severe. Rectal biopsy Imperforate Anus Imperforate anus is usually diagnosed in the neonatal nursery. Passage of meconium is delayed or is noted to take place through an abnormal location as a result of the presence of a fistula. Treatment is surgical; the actual procedure depends on the level and the extent of the defect. Anorectal manometry Distention of the rectum causes relaxation of the internal sphincter *Note that ultrashort-segment Hirschsprung disease may have clinical features of functional (acquired) megacolon. Spina Bifida and Spina Bifida Occulta Defecation disturbances, most frequently constipation, are common in patients with spina bifida and spina bifida occulta, especially if the defect involves the lumbosacral spine. The spinal and nerve root defects result in poor functioning of the terminal bowel. Voluntary external sphincter control and rectoanal sensation are most often diminished or absent, and the degree of difficulty with defecation is related to the degree and the extent of the injury. Most patients can achieve an acceptable level of continence via an individualized bowel regimen. Dietary fiber, stool softeners, suppositories, and enema continence catheters are treatment options. Treatment of patients with spinal or nerve injury or dysfunction from other causes is similar. Therapy for these complications includes correcting electrolyte abnormalities (hypokalemia), broad-spectrum parenteral antibiotics, bowel rest, rectal tube placement, and if needed, emergency cecostomy or colectomy. Treatment for Hirschsprung disease is surgical resection of the affected segment of bowel and various strategies for an ileal or colonic rectal pullthrough procedure. Chronic Intestinal Pseudoobstruction this disorder is characterized by manifestations of intestinal obstruction without an identifiable anatomic lesion and may be secondary to an intestinal neuropathy or myopathy. Congenital cases may be sporadic or inherited, in either autosomal dominant or recessive patterns. Manifestations include abdominal distention, emesis, constipation, growth Metabolic Diseases the appropriate laboratory tests should be performed to rule out the various metabolic and endocrinologic conditions that may manifest with constipation (see Table 16. As the fecal mass accumulates, it causes rectal distention, increases rectal compliance, and eventually results in blunted or absent sensitivity of the rectum to the presence of liquid stool passing around a firm fecal mass. Children with encopresis usually pass small stools and do not completely empty the rectum. It is important to specifically question the patient or parents regarding these massive stools because this information is frequently not volunteered. Encopresis has been incorrectly considered a symptom or manifestation of psychiatric illness. It was thought that the patient retained stools either consciously or subconsciously as a way to rebel against, please, or anger caretakers. Although encopresis may be seen in association with emotional and behavioral problems, it is usually the result of painful defecation followed by a pattern of stool withholding, leading to chronic constipation, overflow encopresis, and possibly poor relations with peers as a result of fecal soiling. In the few patients in whom encopresis is truly a manifestation of psychiatric disease, there is often no stool retention, and the prognosis for fecal continence with therapy is poor. Idiopathic constipation with or without encopresis may compress the bladder by a dilated rectum, thus causing stasis and urinary tract infections. In routine neonatal screening, hypothyroidism manifesting solely with constipation is rarely seen. It should be suspected in any infant presenting with constipation and a history of prolonged neonatal jaundice. Neurologic Disease Children with neurologic disease may have constipation for many reasons, including poor intestinal motility, lack of dietary fiber, and poor awareness of rectal vault distention with stool retention. Any illness affecting the spinal cord or sacral nerves, degenerative muscle diseases, cerebral palsy, and demyelinating diseases can result in constipation. Medication-Related Constipation A complete medication history may reveal substances that can cause constipation (see Table 16. Long-standing constipation leads to encopresis, the deposition of stools in the undergarments or other unorthodox locations that persists or occurs beyond the age that is considered culturally appropriate for achieving continence. A detailed history of bowel patterns identifies many children with normal bowel movements whose parents need reassurance. The history should include a review of all medications and a search for an associated chronic disease, such as a metabolic or neurologic disease. This complete history, combined with a careful physical examination, including the spine and sacral area, the location of the anus, and a digital rectal examination, should alert the physician to the need for further evaluation. Red flags include onset in the neonatal period, growth failure, and prolonged jaundice in the neonatal period. Distinguishing features associated with Hirschsprung disease are listed in Table 16. The anal position index: a simple method to define the normal position of the anus in the neonate. Urological manifestations associated with chronic intestinal pseudo-obstructions in children. Comparison of breast- and formulafed normal newborns in time to first stool and urine. Densmore An abdominal mass or abdominal fullness in a child usually becomes apparent when it enlarges enough to be visualized during bathing or palpable on physical examination. Masses may arise from intraperitoneal, retroperitoneal, or abdominal wall locations and emanate from both solid and hollow viscera. Hepatomegaly and splenomegaly often represent systemic illnesses such as infection, hemolysis, storage disease, or malignancy. A child with an abdominal mass requires a prompt and thorough work-up with testing guided by history, physical examination findings, age, and gender. Early surgical referral may assist in this work-up following a directed screening approach. The duration and character of associated symptoms are important for narrowing the differential diagnosis. A history of abdominal trauma should be elicited, as solid organ injuries may result in hematoma, seroma, persistent pseudocyst, or arteriovenous malformation.
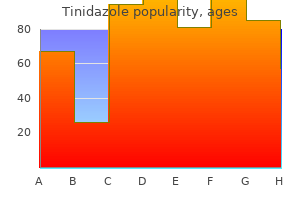
Cheap 500mg tinidazole free shipping
Tics often become worse during stress but may improve during activities requiring moderate physical or mental activity virus hives buy tinidazole uk. Tics need to be differentiated from other abnormal movements, such as chorea, athetosis, dystonia, myoclonus, and hemiballismus, which may be associated with an underlying neurologic condition or may be medication-induced. Complex motor tics are repetitive movements of several muscle groups in coordination, such as repetitive grooming behaviors, deep knee bends, or smelling of objects. Simple vocal tics are defined as nonverbal noises, such as throat clearing or grunting sounds, whereas complex vocal tics are intelligible words. Complex vocal tics may manifest as coprolalia, the repetitive, stereotyped vocalization of obscenities. Chronic motor or vocal tic disorder: either motor or vocal tics lasting longer than a year 3. Tourette disorder: both motor and vocal tics lasting longer than a year Tourette disorder consists of multiple motor and vocal tics of at least 1 year in duration. In some families, this illness is inherited as an autosomal dominant condition, with 70% penetrance in females and near complete penetrance in males. The median age at presentation is 7 years, though some children may present as early as 2 years. While coprolalia is popularly thought to be a common feature of Tourette disorder, fewer than 10% of affected patients have this form of complex vocal tics. Multiple motor and vocal tics lasting longer than a year with no tic-free intervals longer than 3 months 2. No medical cause for the tics Tics may lead to the patient being socially ostracized. Severe recurrent temper outbursts that manifest with verbal or behavioral aggression out of proportion to the situation in intensity or duration 2. Rates are higher in males and school-aged children than in females and adolescents. The hallmark of a substance use disorder is the continued use of a substance despite it causing ongoing negative cognitive, behavioral, and physiologic symptoms. The other hallmark of substance use disorder is the significance of the negative behaviors, such as verbal or physical aggression, defiance, lying, or stealing. Additional symptoms of depression may include significant weight changes, sleep dysfunction, psychomotor agitation or retardation, fatigue or loss of energy, feelings of worthlessness or guilt, diminished concentration, and suicidal ideation or thoughts of death. At least 3 of the following symptoms must be present if the mood is elevated or expansive (4 symptoms are required if the mood is irritable): inflated self-esteem or grandiosity, decreased need for sleep, pressured speech, flight of ideas, distractibility, increased goal-directed activities or psychomotor agitation, and excessive involvement in pleasurable activities with a high potential for painful consequences. Other criteria required for diagnosis are identical to that of a manic episode except that the symptoms are not so severe as to cause marked impairment in social or occupational functioning, hospitalization is not required, and no psychotic symptoms are present. Affected individuals exhibit at least 4 of the following behaviors in a consistent manner over a 6-month period: 1. In prepubertal children, it occurs more frequently in boys; however, in adolescents, its incidence is equal in both sexes. Affected preschool-aged children sometimes exhibit increased motor activity, difficulty being comforted, and overreacting to situations. Affected school-aged children have low selfesteem and a low tolerance for frustration. In addition, these patients may be at increased risk for conduct disorder, antisocial personality disorder as adults, substance abuse, major depressive disorder, and suicide. A child has conduct disorder if he or she has repetitively violated the rights of others and of society. Aggression toward people or animals, such as intimidation, initiation of fights, use of weapons, cruelty to people, cruelty to animals, rape, confrontational theft or mugging 2. Deceitfulness or theft, such as breaking into houses or cars or stealing items of nontrivial value 4. Serious violation of rules, such as curfew violation, running away, or truancy before the age of 13 years (for running away to qualify as a symptom, it must occur twice, or once if it was lengthy, and must not be an attempt to escape sexual or physical abuse) Conduct disorder is classified as childhood onset if symptoms occur before 10 years of age and adolescent onset if symptoms occur at or after 10 years of age. Children initially present with lying, initiating fights, and truancy; as they get older, they progress to more violent acts. Boys are more likely to exhibit acts of violence, such as fighting and stealing, than are girls, who are more likely to exhibit truancy, runaway behavior, and high-risk sexual activity. Half of these children may develop antisocial personality disorder, which is a severe conduct disorder of adulthood that is usually associated with criminal activity. The earlier the onset of conduct disorder, the greater the risk of developing antisocial personality disorder as an adult. Although the cause of conduct disorder is unknown, both genetic and psychosocial factors play a role. A history of parental rejection, difficult infant temperament, physical or sexual abuse, early institutional living, and lack of appropriate discipline are associated with the development of conduct disorder. A biochemical or genetic cause for this condition has been postulated due to the high prevalence of this condition in families with psychiatric disorders. When assessing mood disturbances, it is essential to screen for symptoms suggestive of bipolar illness as these patients have a risk of becoming manic when treated with antidepressants. The evaluation of any patient with a disruption in mood should include an assessment of the risk of suicide. Major depressive disorder is associated with serious risks of both suicide and significant social and academic impairment (see Table 27. Even though a child may be pervasively sad, he or she may also present with behavior problems and irritability. The psychotic symptoms are typically mood-congruent auditory hallucinations and delusions of guilt, medical illnesses, or deserving punishment. During this period, the patient has to have at least 5 of the following symptoms: 1. Feelings of worthlessness or guilt Are there symptoms of mania or hypomania (abnormally elevated mood) Psychomotor agitation or retardation these symptoms should not be secondary to bereavement, medical conditions, substance abuse, or bipolar disorders. The clinician must decide whether the reaction to the stressor is normal, an adjustment disorder, or major depression. There is also a 3-fold increase in major depression in children who have a parent with depression. The differential diagnosis of major depression encompasses various medical disorders, including neurologic disorders, endocrine disorders such as hypothyroidism or hyperparathyroidism, side effects from medications such as H2-blockers or isotretinoin, and substance abuse or use (see Table 27. Major depressive disorder can manifest at any age; however, most patients present in early adulthood. Children usually present with somatic complaints, social withdrawal, and irritability, whereas adolescents often present with psychomotor retardation, thoughts of guilt and worthlessness, and excessive sleep. Approximately 15% of children with major depression eventually develop bipolar disorders. Fifty percent of children with major depression have multiple episodes, frequently associated with significant stressors. Approximately 25% of patients with certain chronic medical conditions such as cancer or diabetes develop major depressive disorder during the course of the illness. The main difficulty in diagnosing major depression is that the gravity of the depressive mood is often not always apparent to the parent and the clinician. Given that children and adolescents often present with irritability, sullenness, or mean-spiritedness, the parents and/or clinician may attribute this behavior to typical adolescent behavior. These children do not always appear sad and the clinician should have a high index of suspicion of major depression in any child who presents with sullenness and irritability. Obtain a thorough family history for symptoms and formal diagnoses of mood disorders. In the majority of cycles, at least 4 of the following symptoms: marked affective lability (mood swings, increased sensitivity to rejection), irritability or anger, increased interpersonal conflicts, 427 depressed mood, feelings of hopelessness or self-deprecating thoughts, anxiety, tension 2. At least 1 of the following: decreased interest in activities, difficulty concentrating, lack of energy, change in appetite, change in sleep, sense of being out of control, physical symptoms of breast tenderness, joint pain, bloating, or weight gain 3. Symptoms present during the majority of cycles over the year prior the severity of symptoms is similar to that in other psychiatric disorders, such as major depression or generalized anxiety disorder, though the duration of symptoms is shorter. Nonetheless, symptoms do need to be severe and cause marked impairment in functioning in order to satisfy diagnostic criteria.
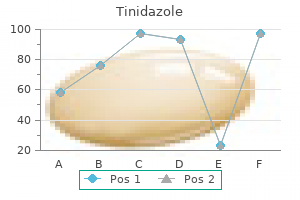
Purchase tinidazole australia
In neonatal infections treatment for dogs broken toe cheap 300 mg tinidazole with amex, tests measuring IgM have become more sensitive and specific. Antigen tests and cultures that grow the parasite are also available but primarily on an investigational level. Syphilis, caused by the spirochete Treponema pallidum, is common in the United States (see Chapter 18). In primary syphilis, in which the inoculation site is usually the genital area, regional lymphadenopathy with painless, firm nodes occurs at the time that a chancre is observed. Thus, inguinal adenopathy in an adolescent who is sexually active mandates further examination and work-up for sexually transmitted infections such as syphilis. In secondary syphilis, the organism has disseminated, causing multiple organs to be involved. Lymphadenopathy, regional or generalized, is common and often includes epitrochlear nodes. Syphilis therefore should be at the top of the differential diagnosis in sexually active adolescents with rash and lymphadenopathy. Pregnant women with syphilis who are untreated readily transmit the disease to the fetus, causing congenital syphilis, often with significant sequelae. Infants with congenital syphilis may also have generalized lymphadenopathy, although this finding is less common than other systemic symptoms, such as hepatosplenomegaly, snuffles, and periosteal reactive disease. The diagnosis has been complicated by the inability to grow the organism in vitro. Dark-field examination of tissue from chancres or mucous lesions shows numerous spirochetes, but dark-field methods are often unavailable to routine laboratories. Nontreponemal serologic studies rely on host production of antibodies to nonspecific lipoidal host tissue antigens that arise as a result of infection with the spirochete. Levels of these antibodies decline after adequate treatment and are useful in confirming eradication of the infection. False-positive reactions can occur, particularly in individuals with connective tissue disorders or mononucleosis. These antibodies usually remain present for the life of the infected individual, even if the patient receives adequate therapy. Lymphadenopathy is frequently among the presenting findings in patients with leukemia or lymphoma. Enlarged lymph nodes may be noted in an isolated, regional, or generalized distribution, with or without classic systemic symptoms, such as fever, malaise, night sweats, weight loss, and anorexia. Malignant nodes are usually firm, rubbery, fixed, and nontender, and may be matted. Unlike many of the acute lymphadenopathies caused by infectious agents, most lymph nodes that are malignant increase in size gradually. Approximately 50% of children with acute lymphoblastic leukemia have adenopathy at the time of diagnosis. Nodal disease may be either generalized or localized to regional nodal groups, often the cervical chains. Nodal disease is frequently accompanied by other signs and symptoms, including fevers, malaise, weight loss, pallor, bone pain, petechiae and bruising, splenomegaly, or hepatomegaly. The complete blood count usually demonstrates anemia, thrombocytopenia, leukocytosis or leukopenia, circulating blasts, or some combination thereof. Some patients may have normal peripheral blood laboratory results on initial evaluation. Acute myelogenous leukemia is less common in children but may manifest in a similar manner. Bone marrow biopsy and aspiration must be performed, and the findings are diagnostic. Non-Hodgkin lymphoma is a relatively common childhood malignancy and often manifests with mediastinal or pleural disease. Adenopathy in the supraclavicular, cervical, or axillary regions is usually present and may occur in the absence of chest involvement. Because lymphoblastic lymphoma may represent a variant of acute lymphoblastic leukemia, the signs and symptoms of leukemia and lymphoma may merge. Non-Hodgkin lymphoma of B cell origin (Burkitt and nonBurkitt lymphoma) in children in the United States usually originates in an intraabdominal site, and regional adenopathy, if present, is then in the inguinal or iliac regions. Hodgkin disease often manifests with painless cervical or supraclavicular lymphadenopathy in older school-aged children and adolescents. Nodes are firmer than those seen in patients whose nodes are enlarged in reaction to infections. In a small number of children with Hodgkin disease, the size of the nodes may wax and wane for several months before a definitive diagnosis is made. Supraclavicular nodes usually indicate intrathoracic disease, which is present in 60-70% of patients at the time of diagnosis. Approximately 30% of patients with Hodgkin disease have systemic symptoms at presentation, including fatigue, weight loss, fevers, night sweats, and poor appetite. Some patients with Hodgkin disease also have unusual symptoms, such as pruritus, hemolytic anemia, and chest pain after alcohol ingestion. Such systemic symptoms with lymphadenopathy are red flags for immediate work-up for malignancy. Diagnosis is confirmed by biopsy of involved nodes and/or bone marrow aspiration, if the tumor has spread to the bone marrow. Disseminated neuroblastoma may manifest as diffuse adenopathy in younger children. Nodes range in size from 1-3 cm, are painful or tender in only 50% of cases, may be multiple, and must be differentiated from lymphoma. Node involvement may occasionally be bilateral or present in axillary or supraclavicular regions. The etiology is unknown, but evidence supports an abnormal immune response; the diagnosis is made by lymph node biopsy. Histologic features include necrosis with karyorrhexis, a histiocytic infiltrate, crescentic plasmacytoid monocytes, and an absence of neutrophils. The disease is self-limiting and usually spontaneously resolves within 6 months, although relapses may occur. Many autoimmune diseases have been associated with Kikuchi-Fujimoto disease, most commonly systemic lupus erythematosus. Patients have massive bilateral painless and mobile cervical lymphadenopathy with associated fever, leukocytosis, and elevated erythrocyte sedimentation rate; polyclonal elevation of IgG may be present. Other nodal chains may be involved; extranodal involvement occurs in 40% of cases. The most common sites are the skin, followed by the nasal cavity and sinuses, palate, orbit, bone, and central nervous system. Histology demonstrates pale histiocytes containing engulfed lymphocytes, and immunoreactivity to S100 protein in large histiocytes, in conjunction with expected clinical features, is diagnostic. Castleman disease is an uncommon lymphoproliferative disease that is also called angiofollicular lymph node hyperplasia. Enlargement of a single node, most often in the mediastinum or abdomen, is the most common localized presentation. Other tumors, such as rhabdomyosarcoma and thyroid cancer, manifest in rare cases with lymphadenopathy caused by local or disseminated metastasis. Ulceroglandular (lymphocutaneous) disorders usually involve an initial injury or bite to an extremity with a resulting cutaneous lesion (ulcer, eschar, papule) and enlarged regional nodes, with or without lymphangitis (Table 36. In some, the cutaneous lesion is secondary to hematogenous spread; in these circumstances the lymph node enlargement may then be generalized (monkeypox). Asymptomatic, unilateral, chronic, cervical lymphadenopathy in Asian boys is suspect for Kimura disease. This benign condition is characterized by peripheral eosinophilia and elevated immunoglobulin E levels. Histologic specimens show a massive eosinophilic infiltration of the nodal architecture. This benign condition necessitates no therapy unless the adenopathy creates functional disability.
Purchase 1000mg tinidazole with amex
Endocrinologic disturbances antibiotic resistance education purchase tinidazole with visa, such as hypothyroidism, can be associated with constipation. A total IgA level should be performed concomitantly to exclude IgA deficiency confounding interpretation. Plain films of the abdomen are rarely necessary, although if obtained may demonstrate stool in the large bowel. This information is occasionally useful in the case of a child with complaints of diarrhea whose symptoms are due to fecal impaction and overflow of liquid stools. Suspected Hirschsprung disease is evaluated radiographically via a liquid contrast enema. Neurologic symptoms are evaluated with magnetic resonance imaging of the spine and/or brain, particularly to assess for evidence of spinal dysraphism or a tethered cord. Concerns for intestinal dysmotility may be evaluated with a Sitz marker study, in which the patient swallows a capsule of radiopaque circular markers and undergoes serial abdominal radiographs to determine the distribution of the markers. After 3 days, any markers that have not yet been evacuated in the stool should be located in the rectum. The presence of markers more proximally in the gastrointestinal tract suggests enteric nervous system or neuromuscular pathology. A rectal motility evaluation may be helpful in the diagnosis and management of chronic constipation. Anorectal manometry can be used to evaluate the integrity of the muscles and the innervation of the defecatory mechanism. Determining the sensory threshold provides valuable diagnostic information: patients who cannot detect a balloon filled with 120 mL of air usually have encopresis. Hirschsprung disease is unlikely if reflexive relaxation of the internal anal sphincter occurs in the presence of rectal distention. Manometry and electromyography document the presence of paradoxical contraction of the external anal sphincter on attempted defecation. Anorectal manometry can also be used as a therapeutic modality in biofeedback therapy in patients with constipation and encopresis and in patients with paradoxical external anal sphincter contraction. Total colonic motility is performed by placing a catheter in the colon to monitor pressures from the rectum to the cecum. Motility tracings reveal information about the function of the colon that is useful in diagnosis and treatment, particularly if surgical options need to be explored. An algorithmic approach to the evaluation of pediatric constipation is presented in. Hirschsprung Disease Congenital aganglionic megacolon, or Hirschsprung disease, is a common cause of neonatal intestinal obstruction, occurring in approximately 1: 5000-1: 15,000 live births, with a male-to-female ratio of about 4: 1. The disease is rare in premature births, may be familial, and is associated with trisomy 21, Waardenburg syndrome, multiple endocrine neoplasia type 2A syndrome, and piebaldism. The absence of ganglion cells in both the Meissner (submucosal) plexus and the Auerbach (myenteric) plexus results in an inability of the involved segment of bowel to relax in response to distention from the presence of stool. Diagnosis may be delayed into childhood and, in rare cases, into adolescence or even adulthood in some patients with ultrashortsegment disease. Differentiating Hirschsprung disease from functional constipation may be challenging in older patients or in those with short-segment disease (Table 16. Other conditions that may mimic Hirschsprung disease include other abnormalities of intestinal innervation such as chronic intestinal pseudoobstruction and hyperganglionosis. The lesion begins at the internal anal sphincter and extends continuously into the rectum or the rectosigmoid in 75-80% of cases. In 10% of cases, there is total colonic aganglionosis; in another 10%, there is variable involvement of the small intestine in addition to total colonic disease. Delayed passage of meconium is the most common manifestation in the neonate, followed by lower intestinal obstruction (distention, bile-stained emesis), obstipation (no stools), failure to thrive, or, in rare cases, intestinal perforation. In addition, if stool is passed immediately after a rectal examination is performed in an obstipated or constipated patient, Hirschsprung disease should be suspected. A plain abdominal film occasionally reveals distention of the normally innervated bowel proximal to the affected segment. The most useful radiographic test, though, is a liquid contrast enema, which may demonstrate a small-caliber rectum with a transition in the rectosigmoid to the dilated, obstructed, normal proximal colon. Patients undergoing contrast enema should not undergo preparatory bowel evacuation procedures, which decrease test specificity, particularly in short-segment disease. A delayed lateral radiograph performed 24 hours after the barium enema aids in identifying a transition zone in the sigmoid colon. Infectious disease may have sequelae of cyst, lymphadenopathy, or intraabdominal abscess. A family and sexual history are also pertinent, particularly in adolescent females. Modern prenatal imaging frequently identifies congenital malformations and neoplasms, requiring postnatal imaging and surgical assessment. Physical Examination A complete physical exam should be performed in children with abdominal masses. Attention should be paid to the general condition of the child and to signs of metastatic disease. Enlarged lymph nodes and their locations should be noted, the skin inspected, and the lungs and heart auscultated. Extremities should be evaluated for evidence of swelling, venous phlegmasia, or evidence of embolic disease. Genitourinary exam should make note of any inappropriate virilization, testicular changes, and hymenal patency in the case of a female with a low pelvic mass. In addition, a neurologic examination may reveal signs of nervous system involvement. The eyes should be carefully inspected for periorbital ecchymosis, proptosis, squint, opsoclonusmyoclonus syndrome, heterochromia of the iris, Horner syndrome, and scleral icterus. To successfully perform abdominal palpation in a child, the physician must approach the patient calmly and gently, as the most reliable exams are completed in cooperative and relaxed children. Creative play is sometimes necessary, with the use of pacifiers or bottles to distract the child from the exam. With the patient in the supine position, the symmetry of the abdomen should be inspected, and any visible masses or the presence of ascites should be noted. A very enlarged spleen is frequently visible, with fullness of the left side of the abdomen. The presence of tense fluid-filled hernias or prominent periumbilical veins as sequelae of portal hypertension should be noted. The mass should be localized, and its size, shape, texture, mobility, tenderness, and relation to midline noted. Dull visceral pain conducted by slow C nerve fibers may be reported for inflammatory processes in the vascular distributions of the celiac, superior mesenteric, and inferior mesenteric arteries and referred to the epigastrium, umbilical region, or hypogastrium, respectively. When the inflamed process contacts the peritoneum, peritoneal fast A nerve fibers allow discrete localization of sharp pain to the abdominal wall. Ultrasound is often a very useful adjunct in the evaluation of an abdominal mass and is often available at the bedside. Approximately half of abdominal masses in older children are caused by enlargement of the liver or spleen, or both. The liver is normally palpated in the right upper quadrant and epigastrium extending 1-2 cm below the costal margin. The inferior hepatic margin may be palpated in a thin child, is usually nontender, and moves with respiration. Detection of liver edge by auscultation using skin scratches has been proven unreliable and has been supplanted by the use of readily available ultrasound. The spleen is located in the left upper quadrant and is nonpalpable in most healthy children. The spleen has a rounded tip and should move downward with inspiration and is more superficial than a renal mass. It is equally important for the examiner to palpate the spleen as it is for the spleen to "touch" the examiner during its descent with inspiration.
Hempseed (Marijuana). Tinidazole.
- How does Marijuana work?
- Are there safety concerns?
- Treating increased pressure in the eyes (glaucoma).
- Dosing considerations for Marijuana.
- Treating multiple sclerosis (MS).
- What is Marijuana?
- Dandruff, hemorrhoids, obesity, asthma, urinary infections, leprosy, preventing rejection after kidney transplants, and other conditions.
- Stimulating appetite in people with AIDS.
- Are there any interactions with medications?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96910
Purchase tinidazole with amex
The clinician should seek symptoms suggesting multisystem disease using topical antibiotics for acne discount tinidazole 1000 mg, such as myalgias, arthralgias, headache, precordial pain or pain with inspiration, abdominal pain, jaundice, skin photosensitivity, peripheral edema, alopecia, Raynaud phenomenon, and hematuria. In patients with symptoms that indicate the presence of multisystem disease, a thorough survey of the functional status of the central, peripheral, and autonomic nervous systems is clinically relevant. Examination the physical examination is used to refine the probability of underlying serious illness, to identify rashes typical of a specific diagnosis, to look for noncutaneous disease manifestations, and to identify if further testing or treatment is indicated (Table 40. Lethargy, irritability, altered mental status, decreased activity, poor perfusion, pallor, or cyanosis indicate serious illness; resuscitation and treatment directed at the most likely diagnoses should be initiated without delay. The importance of the height of fever in predicting the risk of serious illness is unclear. Underlying chronic illness and degree of toxicity are more useful for risk assessment than fever height. The presence of tachycardia and tachypnea in any patient with fever and rash suggests the possibility of sepsis. Evidence of alteration in mental status suggests either sepsis associated with decreased organ perfusion or primary meningoencephalitis. The clinical characteristics of the rash are often helpful in establishing an etiologic diagnosis. An exanthem is defined as a skin eruption occurring as a sign of a generalized disease. An enanthem is an eruption on the mucous membranes that occurs in the context of generalized disease. Exanthems and enanthems may be macular, maculopapular, vesicular, urticarial, petechial, or diffusely erythematous. However, morphology may vary as rashes evolve; the rash of Rocky Mountain spotted fever is classically described as petechial, but it may initially be maculopapular. Because a wide variety of infectious agents, including viruses, bacteria, and the rickettsiae, as well as drugs and inflammatory conditions can cause exanthems and enanthems, few of these eruptions are pathognomonic (Tables 40. Specific Skin Lesions Maculopapular Eruptions Macules are flat, nonpalpable circumscribed lesions, while papules are <1 cm, circumscribed palpable lesions. Maculopapular lesions may coalesce into a more confluent morbilliform (measles-like) eruption. A rash with multiple small papules that feels like sandpaper is described as scarlatiniform. Maculopapular rashes are usually seen in viral illnesses, drug eruptions, and immune complex-mediated disorders. The classic childhood exanthems such as measles, rubella, erythema infectiosum (fifth disease, caused by parvovirus B19), and roseola (exanthem subitum, caused by human herpesvirus types 6 and 7) produce a maculopapular rash and are usually clinically recognizable. As with most viral exanthems, the rash starts on the trunk and spreads to the extremities. Examples of causative agents include aminopenicillins, cephalosporins, antiepileptics, and sulfonamides. Many of the hereditary periodic fever syndromes manifest with maculopapular (or urticarial) rash associated with fever (see Chapter 41). Erythema migrans, the distinctive rash of Lyme disease (caused by the tick-borne spirochete Borrelia burgdorferi), begins as a papule at the site of a recent tick bite and slowly expands over days to weeks to form an erythematous, annular lesion, sometimes with partial central clearing. Erythema marginatum, a rare but major manifestation of acute rheumatic fever, is also distinctive. A, Fine pink macules and thin papules becoming confluent on the posterior upper arm, which is a dependent area in this hospitalized patient. B, More edematous ("urticarial") pink papules; unlike true urticaria, these lesions are not transient. Petechiae are pinpoint lesions (<3 mm), and purpura are larger lesions, and can be either palpable or nonpalpable. While the majority of patients with fever and petechiae have a benign illness, their presence, especially in a child younger than 24 months, is of particular concern. Between 2% and 20% of affected patients have an underlying bacterial infection, and depending on the clinical setting, 0. Other potentially serious infections signaled by a petechial rash and fever include bacterial endocarditis and Rocky Mountain spotted fever. Febrile children may develop petechiae after coughing or vomiting; petechiae in this setting are almost always located in the superior vena cava distribution above the nipple line. Noninfectious causes of fever and petechiae include drug eruptions and acute leukemia. These include infectious diseases associated with organisms with a predilection for vascular endothelium, such as N. Purpura followed by subsequent necrosis of skin is referred to as purpura fulminans, which has been reported after relatively benign infections such as varicella or with more serious disorders (meningococcemia). Discrete, raised purpuric lesions (palpable purpura) distributed predominantly over the buttocks and lower extremities are typical for this disorder. While all febrile patients presenting with diffuse or discrete purpuric lesions must be considered 735 to be at risk for bacteremia. Vesiculobullous Eruptions Vesicular rashes (sharply demarcated, raised lesions containing clear fluid), bullae (vesicles exceeding 1 cm in diameter), or pustules (raised lesions containing cloudy fluid composed of serum and inflammatory cells) may be suggestive of focal or disseminated infection with various pathogens or signal a serious drug reaction. Localized pustules and bullae are usually suggestive of pyodermas caused by Staphylococcus aureus, but pustular lesions distributed on the palms and soles in the context of fever may represent infective emboli with microabscess formation (Janeway lesions), which are often caused by S. Orthopoxviruses such as monkeypox, like smallpox, cause a systemic febrile rash illness with lesions that progress from papules to vesicles to pustules concentrated on the face and extremities. It typically presents within 24 hours after exposure to an offending drug, usually a -lactam or macrolide antibiotic. The clinician evaluating the sexually active patient presenting with asymmetric generalized pustules or vesicopustular lesions should also consider disseminated infection with N. Note the varying stages of development (macules, papules, and vesicles) present at the same time. Red, pink, or plum-colored nodules distributed in a seemingly random manner over the skin surface may represent leukemic infiltrates. The subcutaneous nodules of acute rheumatic fever are usually located over bony extensor surfaces near tendons and are found in <5% of patients with acute rheumatic fever. Erythema nodosum (erythematous and painful nodules usually distributed over the extremities) may be associated with viral infections including hepatitis B and C; bacterial infectious agents, most commonly group A -hemolytic streptococcus; Brucella species; Yersinia species; mycobacterial infections; fungal infections with Candida species, Histoplasma capsulatum, Blastomyces dermatitidis; Cryptococcus neoformans, or Coccidioides immitis; or drug reactions, especially in response to oral contraceptives and sulfonamides. Pyoderma gangrenosum and ecthyma gangrenosum are painful cutaneous ulcerative lesions with an erythematous, raised edge. The lesions usually begin as a papule and break down rapidly with central necrosis. It may be seen in immunocompromised patients with systemic infections with bacterial pathogens, such as Pseudomonas aeruginosa and Stenotrophomonas maltophilia (ecthyma). Localized erythema in the context of acute fever is strongly suggestive of cellulitis, erysipelas, or abscess. The presence of warmth, tenderness, and associated lymphangitis is highly indicative. Organisms causing cellulitis or abscess formation are usually inoculated directly into the skin as a result of trauma. However, bacteremic localization is well described among young children with preseptal or facial cellulitis associated with H. In addition, patients with erythema infectiosum tend to have a maculopapular, lacelike rash over the arms, which may spread to the buttocks and thighs. Patients with dermatomyositis may have localized lilac-colored lesions over the eyelids (heliotrope rash), which may be associated with periorbital edema. Such patients characteristically, but not invariably, have an erythematous, scaly eruption on the face, neck, knees, elbows, and phalanges.
Order 300 mg tinidazole free shipping
Vision impairment (monocular or binocular) acquired after infancy obligates the physician to search for a cause such as a retinal degeneration because some causes are treatable (Tables 32 antibiotic use in animals purchase on line tinidazole. The frequency of examinations is dependent on the findings and progression of the disease. Infants with a birth weight of less than 1500 g are 1st examined 4-6 weeks after birth. Follow-up examinations are performed at regular intervals until the retina is fully vascularized. The choice of treatment depends on the zone and the rate of progression of disease. True leukocoria mandates prompt referral to an ophthalmologist as the causes may threaten either vision and/or life. Retinoblastoma is the most feared cause of leukocoria because of its potential to metastasize and cause death. It is the most common malignant ocular tumor of childhood, with an incidence of about 1/15,000. Less common presentations include periocular inflammation, glaucoma, and proptosis. Imaging is helpful to evaluate for calcifications that occur in retinoblastoma and to help confirm the diagnosis as well as to evaluate for pinealoblastoma and extension of the tumor into the orbit. Referral of a patient with suspected retinoblastoma to an ophthalmologist experienced in its diagnosis and management is critical. In about 1% of cases a parent will have a regressed retinoblastoma or retinocytoma. Treatment options include ophthalmic artery chemosurgery, laser photocoagulation, cryotherapy, intravitreal chemotherapy, systemic chemotherapy, and enulcleation depending on laterality, location, extent of tumor, and vision potential. Murky Murky Varies Varies other clues Improves with pinhole Opacity visible Opacity visible or positive fluorescein Pain Ciliary flush Elevated pressures Pain Steamy cornea Patient ill Painless Abrupt Painless Floaters Cannot see in the eye Carotid or heart disease, migraine Headache History Scintillations Toxins Hypopyon Fundus Appearance Normal Normal Normal Normal Normal Normal Pupil Normal Normal, but red reflex decreased Normal but red reflex decreased Small Disfigured Afferent defect Diffuse retinopathy Papilledema (chronic) Gradual Late Varies Varies Retinal lesions Diagnostic Afferent defect Normal Endophthalmitis Varies Varies Varies Often obscured Varies *Refractive error may be more acute when caused by diabetes mellitus. Many infants with cataracts have leukocoria, but all visually significant cataracts can be detected by careful evaluation of the red reflex. Infants with bilateral, visually significant cataracts may present with visual inattentiveness or nystagmus, signs that significant impairment of vision has already occurred. Most cases of unilateral cataract are idiopathic in origin or associated with other ocular anomalies (persistent hyperplastic primary vitreous, anterior segment dysgenesis). Bilateral cataracts have a known genetic basis in about 60-70% of the cases but this is increasing as novel mutations continue to be described. Cataracts are commonly inherited in an autosomal dominant manner but may be inherited in an autosomal recessive or X-linked pattern. They can be associated with metabolic disease such as galactosemia or Fabry syndrome. Cataracts found to be a result of a metabolic disease may be reversible by removal of the offending agent; galactosemic cataracts are potentially reversible if lactose is eliminated from the diet promptly. Usually these are bilateral but in the case of congenital rubella infection unilateral cataract may occur (Table 32. Other metabolic studies, chromosomal evaluation, and genetic consultation may be indicated. Cataracts in older children may be newly acquired due to a metabolic disease, drug exposure such as steroids, or a manifestation of progressive congenital cataracts. Cataract may rarely be the initial manifestation of diabetes type 1 but can occur as a complication particularly in patients with poor glycemic control. Occasionally cataracts, unilateral or bilateral, are caused by an underlying congenital lens defect, which develops into a visually significant cataract at a later age. Ideally, unilateral cataracts must be removed and amblyopia treatment begun in the 1st or 2nd month of life. Bilateral cataracts judged to be visually significant must be treated in the 1st 3 months of life to facilitate an optimal outcome. Aphakic correction in infants younger than 6-12 months with bilateral cataracts is generally provided with extended-wear contact lenses or spectacles. An intraocular lens may be placed in a child with a traumatic cataract if the intraocular structures that support the lens are intact. Amblyopia must be treated with occlusion of the sound eye, often for several years, until stable visual acuity can be demonstrated. Children who have had cataract surgery must be monitored indefinitely to evaluate for delayed complications such as glaucoma and retinal detachment. Several genes causing primary congenital glaucoma, usually autosomal recessive, have been identified. This presents as a classic triad consisting of blepharospasm, photophobia, and epiphora. The cornea may be cloudy due to edema resulting from elevated intraocular pressure. The cornea also enlarges and the axial length of the eye may increase with elevated intraocular pressure. If glaucoma presents after the age of 5 years, it is known as primary juvenile open-angle glaucoma. The natural history of untreated primary congenital glaucoma is blindness resulting from progressive corneal opacification and optic nerve damage. Treatment is surgical although eye pressure lowering medications such as topical blockers and topical or oral carbonic anhydrase inhibitors may be used as temporizing measures. Even with treatment final visual acuity is worse than 20/50 in more than 50% of the patients. Secondary glaucoma may present similarly to primary glaucoma but is associated with other factors such as ocular trauma, inflammation, prolonged steroid use, or cataract. The differential diagnosis of congenital glaucoma includes conditions that demonstrate corneal opacities (Table 32. Photophobia is observed in infants with corneal trauma, corneal deposits, cystinosis, and inflammation (uveitis). Corneal opacification can also be found in infants with corneal dystrophies, metabolic storage diseases such as mucopolysaccharidoses, and forceps-related obstetric trauma. The diagnosis of primary congenital glaucoma or secondary glaucoma is generally confirmed by performing an examination with the patient under anesthesia. The child needs to be quiet and very cooperative in order to get an accurate intraocular pressure reading and a careful examination of the cornea and anterior segment structures. The diagnosis rests on a constellation of abnormal ocular findings, including elevated intraocular pressure, corneal enlargement, anomalies of the drainage angle, and signs of damage to the optic nerve in conjunction with medical history. Inflammation can involve any or all of these structures, and terms such as iritis, iridocyclitis, choroiditis, and chorioretinitis are used to designate which portion of the uveal tissue is involved. Anatomic location such as anterior, posterior, intermediate, or panuveitis are useful in determining etiology (Table 32. Despite the lack of symptoms, uveitis can cause severe vision loss due to the development of edema or deposition of calcium (band keratopathy) in the cornea or retinal edema. A cataract and/or glaucoma may result from inflammation in the eye or chronic use of steroids used to quiet the inflammation. The pupil may have an irregular shape as a result of adhesions to the underlying lens (posterior synechiae). Because uveitis can be caused by infections, trauma, autoimmune disorders, and may be idiopathic, evaluation of the cause of the uveitis requires a thorough pediatric physical examination as well as supplementary radiologic and laboratory testing. In boys, haplotype testing for human leukocyte antigen B27 may be indicated because of the association between iritis and pauciarticular arthritis that may later evolve into ankylosing spondylitis. The management of iritis in children is the elimination of intraocular inflammation. In some cases of noninfectious uveitis, local treatment with topical corticosteroid drops or periocular corticosteroid injections may control the inflammation. In many cases, local corticosteroids are not sufficient to control chronic uveitis. Short courses of corticosteroids may be used, but corticosteroid-sparing drugs are the 1st-line therapy for long-term use due to the many side effects of corticosteroids. Toxoplasmosis caused by the intracellular parasite Toxoplasma gondii is the most common cause of posterior uveitis in children.
Purchase genuine tinidazole online
As a result antimicrobial gorilla glass order tinidazole 500mg with visa, patients become hypertensive after infancy because of increased sodium retention. Thus, there are deficiencies of glucocorticoids, mineralocorticoids, and potent androgens. This results in a partial deficiency of the enzymes 21-hydroxylase and 17-hydroxylase. Mothers may have virilization during Androgen Exposure: Maternal Source Virilizing maternal tumors. In a minority of cases, the lesion is a benign adrenal adenoma, but the majority are ovarian tumors, particularly androblastomas, luteomas, and Krukenberg tumors. Maternal virilization may be manifested by enlargement of the clitoris, acne, deepening of the voice, decreased lactation, hirsutism, and elevated levels of androgens. In the infant, there is enlargement of the clitoris of varying degrees, often with labial fusion if the tumor produced excess androgen during the 1st trimester. The greatest number of cases has resulted from the use of certain progestational compounds for the treatment of threatened abortion. When gonads are found, they typically contain testicular elements; their development ranges from rudimentary to normal. This form of short-limbed skeletal dysplasia is characterized by anterior bowing of the femur and tibia; small, bladeless scapulae; small thoracic cavities and 11 pairs of ribs, along with malformations of other organs. The gonads appear to be ovaries but histologically may contain elements of both ovaries and testes. Defects in Testicular Development the first step in male differentiation is development of the bipotential gonad into a testis. By contrast, even extreme deletions of the long arm of the Y chromosome (Yq-) have been found in normally developed males, most of whom have short stature and azoospermia. This indicates that the long arm of the Y chromosome normally has genes that prevent these manifestations. In many syndromes in which the testes fail to differentiate, Y chromosomes appear morphologically normal on karyotyping. The constellation of nephropathy with atypical genitalia and bilateral Wilms tumor typify Denys-Drash syndrome. There is onset of proteinuria in infancy that progresses to nephrotic syndrome and end-stage renal failure by 3 years of age, with focal or diffuse mesangial sclerosis being the most consistent histopathologic finding. Wilms tumor usually develops in children younger than 2 years of age and is frequently bilateral. These children have a deletion of 1 copy of chromosome 11p13, which may be visible on karyotype analysis. Affected patients have normal stature as adults and a completely female phenotype at birth, including vagina, uterus, and fallopian tubes. At pubertal age, breast development and menarche fail to occur, and hypergonadotropic hypogonadism is present. Familial cases suggest an X-linked or a sex-limited dominant autosomal transmission. The gonads consist of almost totally undifferentiated streaks despite the presence of a cytogenetically normal Y chromosome. There may be hilar cells in the gonad capable of producing some androgens; accordingly, some virilization, such as clitoral enlargement, may occur at the age of puberty. The streak gonads may undergo neoplastic changes, such as gonadoblastomas and dysgerminomas, and should be removed as soon as the diagnosis is established, regardless of the age of the patient. In this rare syndrome, the external genitalia are slightly atypical but more nearly female. Hypoplasia of the labia; some degree of labioscrotal fusion; a small, clitoris-like phallus; and a perineal urethral opening are present. At the age of puberty, no sexual development occurs and gonadotropin levels are elevated. Affected infants usually develop salt-losing manifestations shortly after birth due to an inability to synthesize biologically potent steroid hormones. Normal pubertal changes in some boys could be explained by the normally present type I 3-hydroxysteroid dehydrogenase present in many peripheral tissues. There is no correlation between degree of salt wasting and degree of phenotypic abnormality. Replacement therapy with adrenal steroids is required from infancy on, and with sex steroids at puberty. In the classic disorder, there is decreased synthesis of cortisol by the adrenals and of sex steroids by the adrenals and gonads. Virilization does not occur at puberty; levels of testosterone are low, and those of gonadotropins are increased. Regression of the testes before the 8th week of gestation results in Swyer syndrome; between the 14th and the 20th week of gestation, it results in the rudimentary testis syndrome; and after the 20th week, it results in anorchia with otherwise normal external genitalia. In bilateral anorchia, sometimes referred to as vanishing testes syndrome, testes are absent, but the male phenotype is complete; it is presumed that fetal testicular function was active during the critical period of genital differentiation but that sometime later it was damaged. Bilateral anorchia in identical twins and unilateral anorchia in identical twins and in siblings suggest a genetic predisposition. Coexistence of anorchia and the gonadal agenesis syndrome in a sibship is evidence for a relationship between the disorders. Deficiency of Testicular Hormone Production Genetic defects have been delineated in the enzymatic steps required for the synthesis of testosterone by the fetal testicular Leydig cells, and a defect in Leydig cell differentiation has also been described. Patients with aplasia or hypoplasia of the Leydig cells usually have a female phenotype, but there may be mild virilization. The gene for the disorder is expressed only in the testes, where it converts androstenedione to testosterone. Most patients are diagnosed at puberty because of virilization and the failure to menstruate. Testosterone levels at puberty may approach normal, presumably as a result of peripheral conversion of androstenedione to testosterone. Cryptorchidism is present in 80% of affected males; during surgery for this or inguinal hernia, the condition is uncovered when a fallopian tube and uterus are found. Testicular function is normal in most, but testicular degeneration has been reported. Smith-Lemli-Opitz syndrome is an autosomal recessive disorder caused by mutations in the sterol 7reductase gene. It is characterized by prenatal and postnatal growth retardation, microcephaly, ptosis, anteverted nares, broad alveolar ridges, syndactyly of the 2nd-3rd toes, and severe mental retardation. Patients with Smith-Lemli-Opitz syndrome may also develop adrenal insufficiency due to inability to produce sufficient steroid hormones. The phenotype most commonly associated with this condition in boys consists of a small phallus, bifid scrotum, urogenital sinus with perineal hypospadias, and a blind vaginal pouch. Testes are in the inguinal canals or labioscrotal folds and are normal histologically. Beard growth is scanty, acne is absent, the prostate is small, and recession of the temporal hairline fails to occur. Familial clusters have been reported from the Dominican Republic, Turkey, Papua New Guinea, Brazil, Mexico, and the Middle East. The disorder is inherited as an autosomal recessive trait but expression is limited to males. In about one third of patients, unilateral or bilateral fallopian tube remnants are found. The testes are usually intraabdominal but may descend into the inguinal canal; they consist largely of seminiferous tubules. At puberty, there is normal development of breasts, and the habitus is female, but menstruation does not occur and sexual hair is absent.
