

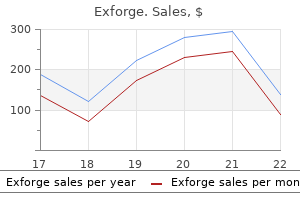
Purchase 80mg exforge free shipping
Comparison of the mutability of triplets shows that some are targeted more often than others blood pressure medication effect on running order exforge amex. For each of the triplets, one can calculate an expected ratio of replacement (R) to silent (S) ChaPtEr 14 Effector Mechanisms In Immunity mutations. For example, in triplets that encode glycine (Gly), six of the nine possible nucleotide substitutions result in an amino acid replacement mutation that gives an R:S value of 2 (6/3). In the case of histidine, eight of nine possible substitutions generate an amino acid exchange, and the ratio is 8 (8:1). The 5 boundary near the promoter is sharp, but the 3 boundary near the enhancer region is less well defined. The I enhancer is the only sequence in the Ig gene cluster that is irreplaceable and must be in the correct orientation. In spite of these preferences, the sequence of the V gene itself does not initiate the mutation, because artificial substrates hypermutate successfully. The precise phase of the cell cycle for introduction of mutations, however, is not yet agreed on, because they have also been detected during the G1 phase. Microarray technology carries our understanding to the last frontier,438 the genes that are involved in initiation and evolution of the reaction. Findings by this approach confirm what is known: the naive, antigenically inexperienced B cell does not divide and does not make Ig but is poised to respond to antigen. The change from naive B cell to centroblast is as striking at the gene level as it is with the techniques that have been used thus far. The centroblast expresses the vast majority of proliferationassociated genes, as well as those that regulate the cell cycle and mitosis. One of the unexpected results was the lack of expression of c-myc proto-oncogene, which is known to be associated with cell proliferation. Studies of gene expression do not detect significant differences between centrocytes and centroblasts, although these two populations have distinct functional characteristics. The light zone contains detectable subpopulations by phenotype or by the expression of transcription factors. In general, the global gene profile of naive B cells is that of suppression, actively maintained by notable inhibitors of signal transduction. The immunoglobulin M of B cells is of low affinity and has been selected before exposure to antigen. The need to improve the affinity is satisfied by another round of B-cell receptor diversification in the germinal centers, after exposure to antigen. It usually places a deoxyalanine (dA) on the sister chromatid, opposite to the abasic lesion (E). The positioning of thymidine (T) opposite dA repairs the lesion, but pol continues over the lesion (translesional pols), altering the bases downstream. As a result, one of the daughter cells is normal (F), whereas the other (with the abasic lesion) has mutational changes (E). However, if the replication fork wins the race to the lesion, the high-fidelity polymerases are stalled. If the polymerase continues its error-prone function, it introduces many more errors, that is, mutations. Memory B Cells Centrocytes can be diverted to the memory B-cell pool by positive signals or by the lack of negative signals. The apoptotic pathway that normally deletes centrocytes belongs to the second category. Apoptosis normally deletes centrocytes that have emerged from the dark zone with nonfunctional or low-affinity receptors. Interference, therefore, with the apoptotic pathway blocks negative selection and results in accumulation of heavily mutated B cells that bring forward a "bad" memory of the primary response. Memory B cells are long-lived and do not secrete antibody until they are challenged again. In such a secondary antigenic challenge, they respond to much smaller doses of antigen, expand clonally, and produce 7 to 10 times more antibody than the antigen-inexperienced B cells. Interaction between idiotypic cells (memory cells for the primary antigen) and antiidiotypic B cells, according to this hypothesis, stimulates enough proliferative activity to maintain the B-cell memory pool. The majority of centrocytes is destined to die, because the mutational changes are detrimental to affinity and avidity. Loss or diminished binding to antigen leads to programmed cell death, and those dead centrocytes are rapidly removed by macrophages, which are now called tingible body macrophages. Some of the centrocytes may be recycled once more through the dark zone for further clonal expansion, diversification, and selection. This recycling provides, once more, the opportunity for reaching the highest affinity peak. In the light zone, the selection process separates the fittest B cells (high affinity) for survival and the unfit (poor or no affinity) for death (negative selection). The new techniques of molecular biology and genomics identify the genes that bring about this spectacular morphologic and functional transformation. B: Immunoglobulin production and expression: B-cell surface versus plasma cell intracellular immunoglobulin. The net result is "activation" of genes for proliferation (1), class switch (2), somatic hypermutation, and signaling B-cell molecules (3). Factors that act as passive repressors interfere with transcriptional activators by competing with their binding site on the promoter or by binding directly to the activation domain of the activator. Type I cytokines consist of four a helices (A, B, C, and D) that are connected by long loops. Depending on the length of the helix, they are distinguished as short chain or long chain. The sequences in helices A and D are the most conserved in evolution, and these helices make contacts with the receptors. Another group of 14 patients of French Canadian descent from Quebec, Canada, had a C-to-T transition in codon 112, resulting in the substitution of Arg with Cys. Later induction of integrins supports T-cell migration and interactions with other cells. The intracellular region has the Box1-, Ser (S)-, acidic (A)-, and proline (H)-rich regions. The a and b subunits are expressed only on lymphocytes, but, after activation, they are also expressed on macrophages. Transition of the cell from G1 to S phase is regulated by G1 cyclins and their associated cdks, which assure the orderly progression from G1 to S phase. The D2-complexes function by phosphorylating their substrate, the retinoblastoma protein, which normally is phosphorylated at a low level and restricts the G1-to-S transition. T-cell activation by antigen induces within the first 30 minutes a number of transcription factors. With the exception of two large deletions, the gc mutations are missense mutations involving one or a few nucleotides. Five mutational hotspots have been identified in exons that encode part of the extracellular domain. The interacting residues are grouped into three clusters,515 with the first two clusters consisting of a nucleus of polar groups that are surrounded by hydrophobic side chains. They have been compared to the avocado fruit, in which a nucleus is enveloped by an oily shell. The N terminal (D1) has the conserved disulfide bonds of an Ig fold, whereas the second domain (D2) has no disulfide bonds; both have unique structural features. Two of them are known as avocado clusters because they consist of a central core of polar groups that is surrounded by a hydrophobic (oily) shell.
Order exforge mastercard
Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport prehypertension youtube buy genuine exforge online. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. A heme export protein is required for red blood cell differentiation and iron homeostasis. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Heme-dependent up-regulation of the alpha-globin gene expression by transcriptional repressor Bach1 in erythroid cells. The relationship between red cell survival and alterations in red cell metabolism. Aceruloplasminemia: molecular characterization of this disorder of iron metabolism. The diagnosis and treatment of iron deficiency and its potential relationship to hair loss. The macrophage: a cellular factory at the interphase between iron and immunity for the control of infections. Immunochemical and mass-spectrometry-based serum hepcidin assays for iron metabolism disorders. Use of reticulocyte cellular indices in the diagnosis and treatment of hematological disorders. Clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. Utility of serum ferritin as a measure of iron deficiency in normal males undergoing repetitive phlebotomy. The definition of anemia: what is the lower limit of normal of the blood hemoglobin concentration Prevalence of anemia in persons 65 years and older in the United States: evidence for a high rate of unexplained anemia. The basis for the mitochondrial iron accumulation in the various sideroblastic anemias can be regarded as either insufficient generation of heme as a result of primary defects in the heme biosynthetic pathway or from faults in mitochondrial functions that involve iron pathways, creating an imbalance between mitochondrial iron import and its utilization. Iron delivery to the erythroid cell does not appear to be downregulated in the face of these alterations, and iron continues to be transported normally to mitochondria, where it accumulates. Very uncommon are several genetically defined syndromic forms involving multiple systems. Acquired sideroblastic anemia is considerably more common than the congenital forms and occurs as a clonal disorder manifesting only anemia or multilineage dysplasia or even myeloproliferative features. Several diverse factors, such as ethanol, certain drugs, copper deficiency, and hypothermia, produce the ring sideroblast abnormality that is fully reversible. Slight hyperbilirubinemia may be noted, as well as an increase in urobilinogen excretion, as a result of a raised erythropoietic component of the "early-label" bilirubin peak. The progeny of surviving ring sideroblasts are often but not always hypochromic and microcytic erythrocytes, a finding that provides morphologic evidence of impaired hemoglobin production as well as an initial clue to the diagnosis. The degree of hypochromia and microcytosis varies considerably from one form of sideroblastic anemia to another. Often, dimorphism is pronounced, with a hypochromic/microcytic population of cells existing side by side with a normal or even a macrocytic one. The siderotic mitochondria of the developing cell may be retained in some circulating erythrocytes (Pappenheimer bodies) and are regularly found with concurrent hypofunction or absence of the spleen; these cells are the nearly pathognomonic siderocytes in the Wright-stained blood smear18. A common feature of many sideroblastic anemias that are not reversible is an excess of total body iron. The serum iron concentration is increased, often to the point of complete saturation of transferrin, and the level of serum ferritin roughly reflects the degree of iron overload. The ineffective erythropoiesis mediates increased intestinal absorption of iron by suppressing hepcidin production. Near that time ironcontaining granules in erythroblasts, including their perinuclear distribution, were described separately,31,32 and 10 years later were demonstrated to represent iron-laden mitochondria. With the advent of molecular biology tools, the genetic causes of several clinically distinct types of congenital sideroblastic anemia and in part their pathogenesis have been elucidated during the last two decades. The expression and regulation of erythroid heme synthesis are unique in that they are linked (a) to the differentiation events after the signaling action of erythropoietin on erythroid precursors when they acquire the machinery for hemoglobin synthesis, (b) to the availability of iron, and (c) to the production of globin during development of the red cell. In this manner, regulation of protoporphyrin production is linked to iron availability and to h //: tp t. For this enzyme, two tissue-specific isozymes are produced by a single gene, which contains two promoter regions, generating housekeeping and erythroid-specific transcripts with alternative first noncoding exons (exons 1A and 1B). To what extent the erythroid-specific enzyme has a regulatory role in the overall production of heme in erythroid cells is not known. Single genes encode these enzymes, and erythroid-specific transcription products are not known for them. However, erythroid-specific regulation of their expression is accommodated by the presence of promoter sequences in their genes for binding of erythroid transcription factors. It may be facilitated by a cytoplasmic chaperone,112 or through direct interaction of endosomes with mitochondria. This heme exporter is essential for erythropoiesis as its loss leads to heme accumulation in the mitochondrion and termination of erythroid differentiation. The events coordinating the production of globin chains with the rate of heme synthesis are well understood, and appear to occur at more than one level. Moreover, absence of this kinase adversely modifies the phenotype of disorders of heme synthesis (iron deficiency, protoporphyria) and of globin production (b-thalassemia). They are clinically and genetically heterogeneous, with diverse underlying causes, inheritance patterns, phenotypes, and associated features. Genetic analysis and molecular approaches have revealed a spectrum of specific defects, some appearing as isolated anemia and others involving multiple systems3,122,123 (Table 24. Defined underlying defects affect heme biosynthesis directly or indirectly by disrupting Fe-S cluster biogenesis, or they involve pathways related to mitochondrial protein synthesis. Severity of anemia is highly variable within some types, and morphologic features overlap among them. Respiratory chain components Thiamine transporter Normal/ increased Not reported Decreased Increased Increased Not reported Not reported Not reported Increased Normal/increased Severe in time Severe Severe Severe Mild to severe normal or increased in females expressing the disorder. However, in many kindreds, the anemia has occurred only in females and may have been lethal in hemizygous male conceptions. Usually, pharmacologic amounts of pyridoxine are required when an erythropoietic response occurs, but the response is variable and rarely complete. Individuals may present with profound anemia only in adulthood or even late in life,129,140,141,142 suggesting that the disorder can progress with time. In some cases, prior additional dietary or medicinal intake of pyridoxine,143 possible changes in pyridoxine metabolism with advancing age,144 or initiation of hemodialysis145,146 can be factors in unmasking mild phenotypes that were not symptomatic at a younger age. In female patients expressing the disease, skewed X inactivation in hematopoietic tissue that occurs with advancing age147 and involves progressive inactivation of the X chromosome bearing the normal allele has been the explanation as the anemia usually evolved in adulthood135,136 or late in life. However, not infrequently the disorder is milder or asymptomatic and may be discovered only in young adulthood or even in later life. Liver biopsy reveals iron deposition that is indistinguishable from hereditary hemochromatosis36. In the X-linked form the iron burden does not correlate with the severity of anemia, and, not infrequently, well-established but asymptomatic micronodular cirrhosis is discovered in the third or fourth decade. The most dangerous manifestations of the iron overload are cardiac arrhythmias and congestive heart failure, which usually occur late in the disease course. In severely affected infants or young children, growth and development tend to be impaired. The red cell volume distribution is usually abnormally wide and, notably, dimorphism is seen in males with the X-linked form124 as well as in autosomal forms of the disease. Leukocyte and platelet values usually are normal; they may be reduced in the presence of splenomegaly (hypersplenism). Erythroid hyperplasia is found on marrow examination, and maturation is usually normoblastic with poorly developed cytoplasm. Megaloblastic changes may be observed if complicating folate deficiency is present. Marrow reticuloendothelial iron is increased, and the telltale ring sideroblast emblem is prominent in late, nondividing erythroblasts. A reduced serum haptoglobin level is consistent with the ineffective erythropoiesis. From its structural features, the protein is predicated to function as an amino acid transporter.
Diseases
- CDK4 linked melanoma
- Tang Hsi Ryu syndrome
- Deciduous skin
- Resistance to thyroid stimulating hormone
- Aging
- Cogan syndrome
Quality 80mg exforge
Unlike a-chains arteria humeral order genuine exforge online, which are highly unstable and unable to form soluble tetramers, excess g-chains in fetal life and b-chains in extrauterine life associate to form relatively soluble g4 tetramers (Hb Bart) and b4 tetramers (HbH), respectively. The different characteristics of the excess chains in a-thalassemia are of great importance in determining its pathophysiology; but in addition, the functional properties of HbH and Hb Bart and the number of a genes contribute to the difference in severity of a-thalassemia as compared to b-thalassemia syndromes. Red blood cells in a-thalassemia are rigid, as in b-thalassemia, but unlike b-thalassemia, they are hyperhydrated and have red cell membranes that are hyperstable. Membrane skeletal-bound b-globins become partially oxidized, with consequent membrane damage. In vitro studies have shown that entrapment of b-chains in normal red cells does not result in any significant change in membrane protein function or thiol concentrations, but rather produces changes in red cell deformability, Chapter 34 Thalassemias and Related Disorders: Quantitative Disorders of Hemoglobin Synthesis as reported in vivo in patients with HbH disease. These inclusions can be induced in vitro by vital stains, such as brilliant cresyl blue or new methylene blue, and are more common in splenectomized patients. Membrane-bound inclusion bodies perturb the flow velocity during transit through the spleen capillaries, ultimately resulting in mechanical trapping and macrophagic phagocytosis. This has been suggested by morphologic and ferrokinetic studies and by the analysis of plasma levels of the transferrin receptor. Red cell membrane deformability and stability is even more affected in patients with HbH/Hbcs. HbH and, even more, Hb Bart, have a very high oxygen affinity and show no heme/heme interaction or Bohr effect, hence severely reducing their oxygen-carrying capacity. This knowledge is helpful in clinical practice for planning the management of the patients, and in genetic counseling for the prediction of phenotype from genotype in couples at risk. In patients, the differentiation at presentation between thalassemia major and intermedia is essential to design the appropriate treatment. In fact, the prediction of a mild phenotype may avoid unnecessary transfusions and their complications, while the diagnosis of thalassemia major will allow an early start of the transfusion program, thus preventing hypersplenism and the red cell sensitization often associated with a delayed start of red cell administration. Therefore, the presence of factors able to reduce the globin chain imbalance results in a milder form of thalassemia. These factors are the coinheritance of a-thalassemia or of genetic determinants that increase g-chain production and the presence of silent or mild b-thalassemia alleles, associated with a high residual output of b-globin. A mild phenotype may also be determined by coinheritance of genetic determinants associated with g-chain production, mapping outside the b-globin cluster. The ameliorating effect that results from the presence of mild b-thalassemia alleles is less constant. However, the information obtained from extended genetic analysis may be used for planning appropriate management and for providing adequate genetic counseling, and may also reveal potential new targets for therapeutic intervention. As reported above, the wide range of phenotypic manifestations of thalassemia results from the heterogeneity of the primary mutation and from the coinheritance of other globin gene-associated determinants, which may ameliorate or worsen the disease severity. Subjects with this genotype have severe hypochromia, microcytosis, and anemia, and do not present HbH at the electrophoresis. As a consequence of the relative excess of b-chains, individuals with HbH disease produce a variable amount of this abnormal hemoglobin, a tetramer of b-globin chains (b4). The HbH is unstable and precipitates inside the red cells and to some extent in erythroid precursors, causing membrane damage and premature erythrocyte destruction. As reported above, both hemolysis and ineffective erythropoiesis contribute to anemia in HbH disease, but the predominant mechanism is hemolysis. HbH has a much higher oxygen affinity than HbA and this may worsen the severity of anemia in patients with HbH disease. In the neonatal period, subjects with the HbH disease genotype have a consistently elevated Hb Bart (25%), which may still be detected in small amounts in some adults with HbH disease. The syndrome of HbH disease shows a considerable variability in clinical and hematologic severity. The majority of patients have minor disability, a few are severely affected requiring regular blood transfusions, and rare cases of HbH disease have been described with the hydrops fetalis clinical picture (see below). Reticulocytes range between 5% and 10%, and the a:b-globin chain synthesis ratio is markedly reduced, in the order of 0. Hemoglobin electrophoresis at alkaline pH shows a fast-moving band (HbH) in amounts ranging from 1% to 40%. Sometimes, because of the low quantity and the possible loss due to instability in the preparation of the hemolysate, HbH may escape detection. Determination of the a-globin genotype may be useful for prognosis of HbH disease, because the nondeletion forms are more severe than the deletion forms. Anemia is accentuated during pregnancy and may worsen quite dramatically with infections, fever, ingestion of oxidant drugs, aplastic anemia associated with Parvovirus B19, and hypersplenism. A mild phenotype of HbH disease may result from the homozygous state for nondeletional a-thalassemia. Although the phenotype in some cases is closer to that of the homozygous state for a+-thalassemia, the degree of anemia and hypochromia may be more severe. The peripheral blood contains HbA2, A, Hb Constant Spring, and traces of Hb Bart CliniCal and laboRaToRy feaTuRes a-thalassemia: Clinical Forms Despite the large number of different a-thalassemia alleles (overall more than 100) only four hematologic and clinical conditions of increasing severity are recognized: silent carrier, a-thalassemia trait, HbH disease, and Hb Bart hydrops fetalis. This genotype is characterized in the newborn period by a very mild increased percentage (1% to 2%) of Hb Bart, a tetramer of four g-globin chains (g4), which is produced when there is an excess of g-chains in relation to a-chains. However, failure to demonstrate Hb Bart in cord blood does not exclude the silent carrier state. Carriers of nondeletion defects have quite variable hematologic phenotypes ranging from the a-thalassemia trait to the silent carrier state (see above). However, homozygotes for some nondeletional forms of a-thalassemia may have a mild HbH disease. Patients with HbH disease may develop complications including hypersplenism, leg ulcers, gallstones, and abnormal left ventricular dysfunction. Hypersplenism has been reported in 10% of Thai patients with HbH disease, but seems to be rare elsewhere. Some clinicians recommend folic acid supplementation as for other hemolytic anemias. Patients should be advised to avoid oxidant drugs because of the risk of hemolytic crisis. Occasional blood transfusions may be required when the hemoglobin level suddenly drops as a consequence of hemolytic or aplastic crisis. Splenectomy may be indicated in the presence of hypersplenism, but the potential complication of venous thrombosis, reported in some patients with HbH disease following splenectomy, should be considered. A fetus homozygous for a0-thalassemia produces mainly Hb Bart (g4), which is functionally useless for oxygen transport, and his or her survival to late pregnancy is due to the presence of small amounts of embryonic hemoglobins Portland 1 (z2g2) and Portland 2 (z2b2). There is a marked variability in the intrauterine clinical course of fetuses with Hb Bart hydrops fetalis. In some cases pregnancy proceeds to term, but the fetus is stillborn or severely ill; in others the fetus does not become hydropic and is born normally. Complications during pregnancy are common and include severe and mild pre-eclampsia (hypertension, fluid retention with or without proteinuria), polyhydramnios or oligohydramnios (increased or reduced accumulation of amniotic fluid, respectively), and antepartum hemorrhage. Postpartum complications include placenta retention, eclampsia (fits and coma), hemorrhage, anemia, and sepsis. At present there is no effective treatment for the Hb Bart hydrops fetalis syndrome. Early treatment with intrauterine transfusions after noninvasive monitoring by Doppler ultrasonography or in utero hematopoietic stem cell transplantation have been attempted, but may not be justified because of the unknown future risks for infants of severe developmental abnormalities. A few cases of hydrops fetalis have been reported in infants with Patients with myelodysplasia may rarely develop an unusual form of HbH disease characterized by the presence of classic HbH inclusion bodies in red blood cells, often detectable levels of HbH (1% to 57%), and a severe microcytic and hypochromic anemia with anisopoikilocytosis.
Generic 80 mg exforge overnight delivery
When transferrin saturation with iron is less than 16% hypertension headache order exforge uk, the red cell production rate decreases, and hypochromic, microcytic cells are manufactured. In the anemia associated with chronic disorders (Chapter 41), the macrophage iron level is normal or increased, but export of iron from macrophages is downregulated. Together, iron deficiency anemia and the anemia of chronic disease are among the most common causes of anemia. Iron deficiency predominates in children and young women; but it also occurs in older individuals, in whom it may reflect occult bleeding due to underlying pathology. The anemia of chronic disorders is most common among elderly people,90 but it also can occur in younger individuals affected by certain chronic inflammatory states. The differentiation of iron deficiency from the anemia of chronic disease and the differentiation of iron deficiency from thalassemia trait syndromes are both common clinical issues. Significant microcytosis is detected in nearly 3% of all patients who require admission to the hospital. The laboratory evaluation of microcytic anemias focuses on screening hematologic studies, followed by more definitive tests to distinguish iron deficiency anemia, the anemia of chronic disease, hemoglobinopathies, or sideroblastic anemias. A few commonly used studies are described below; others are described elsewhere in this text. Distinguishing typical iron deficiency anemia from the anemia of chronic disorders is usually not difficult. Anemia, hypochromia, and microcytosis are typically more pronounced in iron deficiency, as are the degrees of anisocytosis and poikilocytosis. However, when iron deficiency is early and mild, the morphologic findings in the two conditions may be similar. Measurement of Serum Iron and Iron-binding Capacity Serum iron levels are a measure of the amount of iron bound to transferrin. Transferrin also can be measured directly by immunologic techniques and is available in many laboratories. A limitation in the use of serum iron determinations is the considerable variability in values. Even if technical problems are eliminated, serum iron values in an individual can vary from 10% to 40% within a single day or from day to day,93,94 and many normal subjects demonstrate a predictable diurnal variation, with highest values in the morning and lowest values in the evening. The composite normal range is approximately 70 to 200 mg/dl (13 to 36 mmol/L); however, values vary substantially from one laboratory to another. Values below 16% are noted in association with both iron deficiency and the anemia of chronic disorders. The degree of reduction tends to be greater in iron deficiency than in chronic disorders, but considerable overlap exists between these two conditions. In both children and adults, however, a value of less than 5% is almost certainly due to iron deficiency anemia. The values are low (>16%) in iron deficiency anemia and the anemia of chronic disorders, with considerable overlap. ChaPtEr 22 anemia:General Considerations 20 Chronic disease 15 % of patients Normal 10,000 5000 2500 1000 Iron deficient 10 Serum ferritin (ng/ml) 500 200 100 50 25 10 5 2. Low to normal values are found in chronic disease but may also occur in iron deficiency. In most clinical circumstances, the serum ferritin concentration is proportional to total body iron stores. In contrast to serum iron measurements, ferritin values are not influenced by recent iron therapy. For women, mean values of approximately 35 mg/L are noted, with a usual range of 10 to 200 mg/L, when iron deficiency anemia is excluded. Day-to-day variation averages approximately 15%, mostly because of methodologic factors. In patients with excessive iron stores, values are usually greater than 1,000 mg/L and may reach 10,000 mg/L. This phenomenon complicates the diagnosis of iron deficiency when it coexists with inflammatory disease. In some other illnesses, the serum ferritin level increases because of factors other than augmented iron stores. One such condition is liver disease, in which damage to the hepatic cell can cause the release of intracellular ferritin (nonglycosylated and iron-rich). Measurements of serum ferritin in uncomplicated iron deficiency, inflammation, liver disease, and iron overload. Patients with iron deficiency (total iron-binding capacity >400 mg/100 ml and transferrin saturation below 16% or absent marrow iron or both) (open circles). More often, the specimen is stained by the Prussian blue method, which renders hemosiderin blue. In iron deficiency, marrow hemosiderin is absent; in the anemia of chronic disorders, iron is always present, most often of grade 2 or 3+, but sometimes 4 or 5+. Iron stores are greatly increased (5 to 6+) in thalassemia major and in sideroblastic anemias. Nowadays the marrow assessment of iron is used much less, Gale E, Torrance J, Bothwell T. Chemical analysis has been important in evaluating hemochromatosis and transfusioniron overload, because a disproportionate amount of iron is in parenchymal cells in the form of ferritin, which does not stain with Prussian blue. Currently, however, magnetic resonance imaging using T2* technology is replacing liver biopsy as a noninvasive way to measure liver iron content. A soluble, truncated form of the protein can be detected in plasma, and its immunologic quantification can be useful in detecting iron deficiency and distinguishing it from the anemia of chronic disease. Values in iron deficient subjects are clearly increased, and are typically not increased in uncomplicated anemia of chronic disease. Therefore, the value increases in hemolytic anemia, thalassemia, and polycythemia, and decreases in hypoplastic anemia and renal failure. A rough correlation exists between iron stores and urinary iron excretion after deferoxamine administration. Other Tests to Assess Iron Metabolism Red cell precursors normally synthesize slightly more protoporphyrin than is needed for heme synthesis. When iron is not available for heme synthesis, protoporphyrin accumulates in excess as zinc protoporphyrin. The thalassemias are a group of inherited disorders in which synthesis of one of the normal polypeptide chains of globin is severely deficient (Chapter 34). In mild forms of the disease (thalassemia minor), hypochromia and microcytosis are prominent, whereas anemia is absent or mild. In other thalassemic disorders, including homozygous b-thalassemia (b-thalassemia major) and Hb E b-thalassemia, hypochromic, microcytic anemia is usually quite severe. Heterozygotes for this hemoglobinopathy typically have microcytosis without anemia. In addition, basophilic stippling and target cells tend to be more prominent in thalassemia than in iron deficiency. This characteristic has allowed several different measures for differentiating iron deficiency from thalassemia trait and Hb E disorders. Liver Iron Stores Iron stores can also be estimated by liver biopsy using both histochemical and chemical methods of analysis. Bars indicate the mean (dot) and 95% confidence intervals of values in the three groups. ChaPtEr 22 anemia:General Considerations Homozygous hemoglobinopathies, especially Hb C125 and Hb E,53,126 tend to be microcytic and normochromic, and many target cells are apparent in the blood smear (Chapter 34). Distinguishing homozygous b-thalassemia (b-thalassemia major) from b-thalassemia minor is rarely a problem, because the former is accompanied by signs of hemolysis and ineffective erythropoiesis; there also are characteristic findings on the blood smear, including nucleated red cells, extreme anisocytosis and poikilocytosis, and target cells (Chapter 34). However, it is a common diagnostic problem to distinguish patients with b-thalassemia trait from those with iron deficiency. In almost all cases of b-thalassemia trait, the fraction of Hb A2 is increased, whereas the value for Hb A2 is normal or decreased in iron deficiency. Hb H disease is caused by the deletion of three a-globin genes, and the consequence of this is a mild to moderate hemolytic anemia.
Buy exforge 80mg line
Eosinophils have the capacity to exert powerful effector functions against mucosal tissues that may contribute to airway hyperresponsiveness pulse pressure damping discount exforge online amex. However, the full capacity of the eosinophil to elaborate cytokines, the precise microenvironment requirements for such synthesis, the intracellular pattern of production and storage, including the timing of its immunomodulatory function during immune responses, remain the subject of intensive investigation. Eosinophils make up approximately 3% of the bone marrow from healthy individuals, of which 37% is fully differentiated, and the remainder is promyelocytes/myelocytes and metamyelocytes. Cytokines derived from eosinophils may lead to autocrine prolongation of eosinophil maturation and survival in tissues. Extracellular matrix proteins have been shown to modulate eosinophil responses to soluble physiologic stimuli. These receptors function to enhance eosinophil responses and are likely to promote the activation of tissue eosinophils by cytokines and other signaling molecules. Tyrosine phosphorylation enhances the expression of the antiapoptotic protein Bcl-xL in eosinophils, and decreases translocation of the proapoptotic signaling molecule Bax, resulting in decreased activation of apoptotic signaling through the caspase family. Thus, the growth, maturation, and prolongation of survival of eosinophils in extramedullary tissues may occur in sites other than the bone marrow. The half-life of eosinophils in the circulation is approximately 18 hours with a mean blood transit time of 26 hours,231 although this is extended in eosinophilic conditions, possibly due to the elevation of systemic eosinophil-activating cytokines that promote eosinophil survival. Based on a study of 740 medical students, the normal range of blood eosinophils was shown to be between 0 and 0. Allergy is commonly associated with eosinophilia in the mild range, whereas parasitic infestation is often characterized by a marked eosinophilia. Eosinophils are predominantly tissue cells, and their major target organs for homing in the healthy individual is the gastrointestinal tract (outside of the esophagus), mammary gland, uterus, thymus, and bone marrow. The gastrointestinal tract is the predominant site of homing for tissue eosinophils in healthy humans. Once they enter target tissues, eosinophils do not return to the blood circulation. Eosinophil numbers can remain high in tissues even when peripheral numbers are low, suggesting that their survival is enhanced upon extravasation. Curiously, pathogen-free laboratory animals have no eosinophils in their blood, and tissue eosinophils are scarce, suggesting that the appearance of eosinophils may be environment- or disease-related. There are three different populations of eosinophils that can be characterized based on their intrinsic buoyant density and responsiveness to stimuli. These are the normodense, hypodense, and primed eosinophils, which can be described in both normal and eosinophilic subjects. Each of these populations responds differently to stimuli, which may be related to their stage of maturation. In addition, they may derive from distinct pools of eosinophils that are genetically regulated. The majority of blood eosinophils (>90%) from normal individuals are normodense, which separate out from other leukocytes in the lower interfaces of Percoll or metrizamide discontinuous density gradients. Hypodense eosinophils can be seen in a proportion of eosinophil production and Survival in peripheral tissue Eosinophil development and maturation may also occur in situ in peripheral (extramedullary) sites outside of the bone marrow. In this case, Eo/B precursors are released into the bloodstream directly from the bone marrow to circulate to sites where they specifically transmigrate in response to locally produced cytokines and chemokines. This may provide an alternative mechanism for the persistence or accumulation of tissue eosinophils. Signaling pathway leading from binding of iL-5 to its receptor in the membrane to transcriptional activation in the cell nucleus via the ras-raf1-MeK-erK pathway. Transcriptional activation is proposed to generate antiapoptotic effects in eosinophils. The mechanisms involved in the selective tissue recruitment of eosinophils across the vascular endothelium and into tissues in allergic reactions occur sequentially in four welldefined steps. These include (1) the tethering of the eosinophil to the lumenal surface of the vascular endothelium during normal transport through the blood vessel, (2) rolling of the eosinophil along the lumenal surface of the activated endothelium in a reversible manner, (3) firm adhesion of the eosinophil to endothelial cells, and (4) transmigration of the eosinophil through the endothelium into target tissues. A further, less understood, step in eosinophil trafficking in the tissues is the in situ differentiation of circulating committed Eo/B precursors. Each of these steps is controlled by a complex network of chemotactic factors and adhesion molecules, which collectively direct the movement of the eosinophil into the tissues. Cytokines and chemokines are elaborated by surrounding tissues to modulate the transmigration of eosinophils into tissues. Many of these mechanisms appear to be controlled at the level of the T-cell response to antigen (allergen)-presenting cells, and the subsequent release of cytokines and chemokines which in turn regulate the activity of eosinophils. The presence of low density (or hypodense) eosinophils appears to be a nonspecific phenomenon that occurs in any eosinophilic condition including parasitosis, asthma, allergic rhinitis, idiopathic hypereosinophilic syndrome, and certain malignancies. It was originally thought that the numbers of hypodense cells correlated with the degree of eosinophilia, although this has not been consistently observed. The first is that hypodense eosinophils frequently co-migrate to the same density as neutrophils in metrizamide or Percoll gradients, thus making it difficult to separate these two cell types. However, the surface expression of numerous other receptors does not differ between normodense and hypodense eosinophils, with some populations. It is possible, therefore, that the formation of low-density eosinophils results from the migration of normodense eosinophils from the bone marrow to the circulation, whereupon they become activated by elevated systemic factors. Another scenario may be that the association between hypodensity and activation is coincidental, with the less dense cells being immature. This heterogeneity in eosinophils has not received significant attention in recent years, likely due to changes in purification techniques for eosinophils. Although early studies generated eosinophils purified by density gradients, using Percoll, for example, by far the most common approach in modern studies is negative selection by immunomagnetic bead separation. During chemotaxis, eosinophils may either become activated in response to local inflammation and release mediators, as in asthma and other related conditions, or they accumulate in tissues in the apparent absence of mediator release. Antibodies specific for adhesion molecules have been applied in this system and have identified critical regulatory molecules required for adhesion and transmigration of eosinophils. This interaction is enhanced after the release of inflammatory mediators from these cells as well as neighboring tissues. Once tethered, eosinophils roll until they become stimulated by a chemoattractant stimulus (indicating local inflammation), which induces activation of a4 integrin receptors on the leukocyte. In addition, rolling appears to facilitate the subsequent adherence and transmigration of eosinophils into tissues. These findings underline the importance of cytokine and chemokine cross-talk in the generation of blood eosinophilia and tissue diapedesis. Complement-mediated inflammation, as seen with parasite infection, is associated with the release of C3a and C5a. C3a increases binding of eosinophil to endothelium but does not increase migration; however, C5a increases both adhesion and migration. Eosinophils move through the endothelium by extending lamellipodia in the form of a uropod, thus leading to lamellar motion. Changes in the binding affinity for adhesion molecules and extracellular matrix proteins are thought to contribute to cell movement on a substratum. Other factors are also produced in mucosal tissues, which are moderately or strongly chemotactic for eosinophils. Eosinophils are a feature of allergic and nonallergic asthma,265 and large numbers of eosinophils and eosinophil granule products are found in and around the bronchi in asthma patients. The use of fiberoptic bronchoscopy with biopsy is considered the gold standard for acquiring the best appreciation of eosinophil involvement in asthma. However, whether degranulation from eosinophils is simply the result of eosinophil infiltration, or if it plays an important role in allergic disease, is yet to be determined. Sputum analysis offers a noninvasive technique showing correlation of eosinophil numbers with clinical outcomes. It appeared that airway hyperresponsiveness could develop during allergen challenge even though blood eosinophilia was lost. Despite developing blood eosinophilia, Eo-/- mice show reduced but not abolished tissue eosinophilia 242 with its associated eosinophil-mediated tissue damage following allergen challenge. Therefore, a key event in eosinophilmediated inflammation leading to airway hyperresponsiveness may lie in the persistence of activated eosinophils in the tissue. These responses were restored by reconstitution of eosinophils353 or a combination of eosinophils and antigen-specific T-cells.
Syndromes
- When you bend the neck backwards or walk more than a few yards
- Ultrasound of the head
- Nerve pain or numbness in the arms or legs
- Look at your face, neck, back of the neck, and scalp. It is best to use both a hand mirror and full-length mirror, along with a comb, to see areas of your scalp.
- Bloody sputum
- To have surgery on your heart and you are at high risk for coronary artery disease

Buy 80 mg exforge amex
They are not phagocytic pulse pressure too close order line exforge, but retain the captured antigen for prolonged periods, in the form of immune complexes. They also have been implicated in the generation of memory lymphocytes and the maintenance of antibody production. Cross section of a paracortical cord showing the corridor, outlined by reticular fibers stained by gomoristain. Cords, channels, corridors, and conduits: critical architectural elements facilitating cell interactions in the lymph node cortex. In the lymphatic vessels, they are known as veiled cells because of their morphology. The lymphatic vascular system plays an important role in the regulation of tissue fluid homeostasis, immune surveillance, and growth and maturation of new vessels, as well as maintenance of existing lymphatic vessels, the functions of which are regulated by tyrosine kinases. Vascular growth in the draining lymph node includes the growth of endothelial cells in the high endothelial venules associated with increased entry of cells into the lymph node. The importance of the lymphatic vessels in fluid and cellular homeostasis provides opportunities for developing inhibitors to target tyrosine kinases and thus affect lymphatic vessel functions or even lymphangiogenesis in the chemotherapy of malignancies. Afferent lymphatics pierce the capsule and empty into a subcapsular sinus, which gives rise to cortical sinuses and often runs along the trabeculae through the cortex to the medulla. Electron microscopic studies of the primary follicles detected reticulum cells with little or no evidence of phagocytic activity making contact with neighboring cells by very tight adhesions or desmosomes. Their cytoplasm is electron lucent and contains lysosome-like vesicles and tubular structures extending to the periphery of the cell. In ontogeny the dendritic cell system develops slowly, and as a result during neonatal life the body depends primarily on innate immunity. This movement modifies the structure of the endothelium, evidenced by intravenously injected small thymocytes which trigger migration of small numbers of lymphocytes. ChaPtEr 11 Lymphocytes and Lymphatic Organs extend from the cortex to the medullary cords and eventually exit from the hilum as veins. The venules which give rise to capillaries possess unique endothelial cells that are tall and cuboidal and therefore contrast with the endothelial linings of other venules that usually are low or flat. Induction of inhibitors, such as antagonist antibodies or decoy receptors, may be used in chronic inflammation, autoimmunity, cell death, or cancer, i. Two types of high endothelial cells are distinguished, one having abundant cytoplasm, and their luminal surface is covered by a coat of filamentous and granular material 1. By electron microscopy, on the basis of the content of their polyribosomes, one cell is lighter and one darker and intensely pyroninophilic. Their cytoplasm contains numerous microtubules which radiate from the centriole and multiple dense bodies which probably are related to lysosomes and function as storage sites for glycoproteins used for the formation of the cellular coat. A striking morphologic feature of these cells is their large Golgi complex with many vesicles which are associated with actively secreting cells not seen in endothelial cells. Some lymphocytes (L) are crossing the wall of the high endothelial venule and enter the lymph node. Acid hydrolase activity and nonspecific esterases that are not present in other endothelia have been detected in these cells, as well as a peculiar ability to switch to anaerobic metabolism. The metabolic activity of the endothelial cells is influenced by the number of circulating T lymphocytes. In congenitally athymic mice or in normal mice following neonatal thymectomy or chronic thoracic duct drainage, i. Some evidence also has implicated the dendritic cells as playing a regulatory role in determining the morphology and metabolic activity of the endothelial cells. This crossing is now recognized as an artifact because all lymphocytes that appeared on electron microscopic sections to be surrounded by endothelial cells were in fact outside of these cells when evaluated with tracer studies. In order to avoid confusion with gonadal cells, however, they were renamed centroblasts. The blast is a large cell (larger than 10 mm) and has a nucleus with loose chromatin and a giant reticulated nucleolus that may occupy as much as one half of the nuclear area. The Golgi apparatus is moderately developed and contains many free polyribosomes but only few cisternae of endoplasmic reticulum. As the endoplasmic reticulum becomes more abundant, these cells are known as plasmablasts. Free polyribosomes are still present and the cisternae of the endoplasmic reticulum do not fill the cytoplasm entirely. Antigenic stimulation drives the differentiation of B cells into plasma cells,156 seen in the medullary cords by day 3 or 4 after immunization, as well as the proliferative activity reflected in the increase of pyroninophilic blast cells in the paracortex. The effector cells of both responses are exported to remote sites for effective control of the infection. It provides the microenvironment for the final differentiation of reticulocytes, platelets, and monocytes, and is the reservoir for erythrocytes and granulocytes and removes aged or deformed red blood cells. The spleen is covered by a connective tissue capsule which is several millimeters thick,159 and in humans it contains only a few muscle cells and is not capable of marked contractions. The internal surface of the capsule is the point of origin of an extensive network of trabeculae that divides the organ into communicating compartments, with a sponge-like appearance and spaces containing the parenchyma, or pulp tissue. The capsule is indented on the medial surface where the blood vessels, lymphatics, and nerves enter and leave. Histologic studies show two kinds of parenchyma: one is stained dark blue to purple because of the predominance of small lymphocytes (the white pulp), and the second is red because of the predominance of blood-filled sinuses (the red pulp). Although the spleen is considered a secondary lymphoid organ, it serves several other functions unrelated to the immune system and affects all blood cells throughout their life span. ChaPtEr 11 Lymphocytes and Lymphatic Organs 243 White pulp the white pulp is the main lymphatic tissue of the spleen and surrounds the blood vessels. The splenic artery branches into trabecular arteries (central arteries), muscular vessels that can alter blood flow by their contraction and relaxation, which follow the trabeculae and periodically give off branches. The reticulum is a scaffolding made of reticular fibers secreted from spindle shaped reticular cells, with conspicuous microfilaments in their cytoplasm, a prominent endoplasmic reticulum, and a nucleus which contains finely dispersed chromatin. It provides support for free cells and blood vessels, runs circumferentially around the central artery, and becomes particularly pronounced in the periphery of the white pulp, where the reticular cells extend concentric sheets of membranes160,161. In the human spleen, some branches curve back from the red pulp to form a delicate network around lymphatic follicles located at the periphery of the white pulp. Tightly packed lymphocytes lie between fibers and reticular cells (arrows) that run circumferentially around the artery. The cord below contains bulky reticular cells (asterisks) and fibers (white arrows). The large cell in the lumen of the sinus is probably a macrophage with several thin cytoplasmic strands (arrow). The beaded black lines at the cut edge of the sinus identify its adventitial layer (terminal bead) and its endothelial layer (subterminal bead). They are large with unusually shaped nuclei, poor in heterochromatin, and their cytoplasmic extensions are thin sheets rather than dendrites. On the basis of cell-surface markers, maturation of B cells in the periphery proceeds through three transitional stages. The vessel is supported by cordal reticular cells and fibers and a fenestrated basement membrane consisting of heavy fibrous strands running circumferentially and lighter longitudinal strands. Thus, the circular bands, like the hoops of a barrel, hold the endothelial cells (staves of the barrel) tightly. The segregation of the two major lymphocyte populations within distinct settlements of the secondary lymphoid organs seems to be determined by the nature of their underlying neighbors. Some authors suggest that the marginal sinus may be functionally equivalent to the postcapillary venule, which in rodents is the site at which lymphocytes enter the splenic parenchyma. The lymphocytes entering the marginal sinus have multiple microvilli with which they establish contacts with reticular cells as they find their way toward their microenvironments, and once they reach their destination the microvilli disappear. These cells have distinct phagocytic and morphologic properties which distinguish them from macrophages in other locations of the spleen, including the ability to bind lymphocytes. These cells differ from naive B cells in morphology, phenotype, and red pulp As seen in histologic sections the red pulp consists of sinuses and cords,186,187 which are composed of a meshwork of reticular fibers and cells. The large reticular cell sheets have microfilaments that endow them with the capacity to retract and extend, and thus determine the available space and regulate blood flow. These cordal spaces receive blood directly from the arterial vessels, as the central artery gives off many branches which terminate in slender, straight, nonanastomosing arterioles that enter the cords of the red pulp but not the sinuses. Some of the arterioles divide into arterial capillaries which are enveloped by a sheath of phagocytic cells. These sheaths were called ellipsoids, but now are called periarterial macrophage sheaths, which in the human spleen is not well developed.
Purchase 80 mg exforge with amex
The average amount of blood lost per period is approximately 50 ml prehypertension chest pain safe 80 mg exforge, representing roughly 25 mg of elemental iron. Menstrual flow should be deemed excessive if more than 12 pads are used each period, if clots are passed after the first day, or if the period duration exceeds 7 days. The number of pregnancies and abortions and the interval since the most recent of these are also important, for each represents significant iron loss. The presence or absence of fever must be known; its presence suggests infection, lymphoma, other neoplasm, or collagen vascular disease. Pains in the limbs, paresthesias, and difficulty in walking suggest pernicious anemia. Abnormal color of the urine, suggesting blood or Hb, may signify urinary tract disease or hematologic problems. Bilirubin is not detected in the urine of people with uncomplicated hemolytic anemia ("acholuric Skin and Mucosal Changes Other changes in the integument occur with anemia. Thinning, loss of luster, and early graying of the hair may occur, the last especially in patients with pernicious anemia, in whom it may precede the development of anemia. This finding is especially noticeable in chronic iron deficiency anemia,39 in which the nails may actually become concave instead of convex. Chronic leg ulcers may occur, especially in patients with sickle cell anemia and rarely in those with other hemolytic anemias. When nutritional deficiency is associated with anemia, symmetric dermatitis may develop, fissures may be present at the angles of the mouth, glossitis may occur, and erythematous lesions on the face, neck, hands, or elbows may be found. Neuromuscular Features Headache, vertigo, tinnitus, faintness, scotomata, lack of mental concentration, drowsiness, restlessness, and muscular weakness are common symptoms of severe anemia. Paresthesias are common in pernicious anemia and may be associated with other symptoms and signs of peripheral neuropathy, and more especially with combined system disease. Ophthalmologic Findings A variety of ophthalmologic findings have been observed in anemic patients. Papilledema related solely to anemia has been described,43,44 and it clears when the anemia disappears. Glossitis and atrophy of the papillae of the tongue commonly occur in pernicious anemia and less often in iron deficiency anemia. Painful, ChaPtEr 22 anemia:General Considerations jaundice"), but a darker than normal color may result from the increased excretion of urobilinogen and its conversion to urobilin. Their presence indicates that the disorder producing anemia may also involve platelets or the liver. Alternatively, the anemia itself may be the consequence of blood loss resulting from a disorder of hemostasis. In all instances, the presence or absence of symptoms suggestive of an underlying disease such as chronic renal disease, liver disease, chronic infection, endocrinopathy, or malignancy must be explored. Scleral icterus suggests the presence of hemolytic anemia or ineffective erythropoiesis. Sternal tenderness near the middle or lower third of the sternum, of which the patient may have been unaware, may represent acute expansion of hematopoietic marrow and can be a useful sign in some patients with acute leukemia. Palpation of the liver and spleen and a systemic check for lymphadenopathy can provide clues to infection, lymphoma, leukemia, or metastatic carcinoma. Even when the color of the urine does not suggest blood, the routine urinalysis should be tested for occult blood. A positive reaction may be due to hematuria, hemoglobinuria, or even myoglobinuria. A useful approach entails asking several questions, outlined in the following sections. Specifically, is the anemia associated with thrombocytopenia or abnormalities in white blood cell numbers or the presence of abnormal leukocytes If the answer to this question is yes, consideration must be given to the possibility of bone marrow failure due to aplastic anemia, leukemia, or other malignant marrow disease. Alternatively, pancytopenia can be secondary to peripheral destruction or sequestration of cells as in hypersplenism. In most cases, these disorders can be differentiated by careful review of screening hematologic studies and close attention to the medical history and physical examination. The number of erythrocytes in the circulation at a given time is the result of a dynamic equilibrium between the delivery of red cells into the circulation on the one hand and their destruction or loss from the circulation on the other. The homeostatic mechanisms of the body bring about recovery from anemia by accelerating erythropoiesis, and this response of the normal marrow is brought about through Is anemia associated with other hematologic abnormalities Yes Bone marrow examination to assess for: Leukemia Aplastic anemia Myelodysplasia Myelofibrosis Myelophthisis Megaloblastic anemia No Is there an appropriate reticulocyte response to anemia At maximum stimulation, the bone marrow is capable of producing erythrocytes at six to eight times the normal rate. The reticulocyte count is traditionally measured by microscopic examination of a smear prepared from fresh blood stained with a supravital stain, such as new methylene blue. More recently, automated methods based on flow cytometry have become widely utilized. The automated methods count a larger number of cells, and exhibit a greater degree of reproducibility. An additional correction of this index needs to be made because reticulocytes released under intense erythropoietin stimulation remain in the peripheral blood for more than the usual 1-day survival time of nonstress reticulocytes. There are a number of ways to adjust the reticulocyte count for the degree of anemia (Table 22. While all of these methods have value, the absolute reticulocyte count is traditionally the easiest to estimate. If Anemia Is Associated with a Less Than Appropriate Reticulocyte Response, What Are the Red Cell Indices Anemia with low reticulocytes usually reflects some impairment of normal erythropoiesis, and this can be due to two kinds of defects. Erythropoiesis may be impaired because of a reduction in red cell precursors (hypogenerative). Alternatively, red cell production may be ineffective, a condition characterized by erythroid hyperplasia in the bone marrow, but with the production of essentially nonviable red cells, most of which do not reach circulation. Because of plasma trapping, centrifugal Hct methods overestimate the volume of packed red cells and, therefore, If Anemia Is Associated with Reticulocytosis, Is There Any Evidence for Hemolysis The most characteristic presentation of hemolysis is reticulocytosis with some degree of hyperbilirubinemia as a marker of increased heme catabolism. The evaluation and diagnostic considerations related to hemolytic anemia are complex and are considered separately elsewhere in this chapter (see Approach to Hemolysis). Plasma trapping increases from 1% to 3% with normal blood to as much as 6% in iron deficiency, a consequence of anisocytosis and reduced cell deformability. The large majority of patients in this category have defects in cellular Hb synthesis due to either iron deficiency, thalassemia trait, or Hb E syndromes (see Approach to Microcytic Anemia). Is the Anemia Associated with a Low Reticulocyte Response and Macrocytic Red Blood Cells Many of these disorders are due to megaloblastic anemia resulting in impaired nuclear development, and the formation of other blood cells is also affected (see Approach to Macrocytic Anemia). Is the Anemia Associated with a Low Reticulocyte Response and Normocytic Red Blood Cells Normocytic anemia, low reticulocyte count, and normal bilirubin levels characterize a large number of anemias. The anemia of chronic disease usually is normocytic, although rarely may be slightly microcytic. In these cases, there usually is clinical evidence of a syndrome associated with cytokine activation. The anemia of renal failure is normocytic and largely is due to reduced erythropoietin production. The red cell indices represent mean values and do not reveal any variation that may exist within a population of cells. Examination of the bone marrow is most useful in reticulocytopenic anemias, particularly when there is more than one hematopoietic cell line affected. Both hypoplasia and marrow infiltrative disease due to leukemia, tumor, or granulomas (myelophthisic anemia) may readily be demonstrated in the bone marrow aspirate and biopsy. If the marrow is normocellular except for reduced erythropoiesis, the underlying cause may be red cell aplasia, renal disease, or endocrinopathy. Examination of iron in bone marrow macrophages was traditionally considered the definitive way to demonstrate decreased iron stores. In most cases, however, the diagnosis of iron deficiency can be made by simple blood tests, thus obviating the need for an iron stain of the bone marrow. On the other hand, to make the diagnosis of sideroblastic anemia, a bone marrow examination is necessary to identify ringed sideroblasts. Megaloblastic anemias usually can be recognized by peripheral blood findings, but a marrow examination will confirm the diagnosis. In some anemias with low reticulocyte counts, marrow erythropoiesis surprisingly is quite active. This is referred to as ineffective erythropoiesis, and it occurs when developing red cells are defective and are destroyed before they leave the marrow or shortly thereafter.
Order 80mg exforge overnight delivery
Summary Less operator dependent (scan) than the combined test and probably as accurate; not recommended despite this (see Table 3 blood pressure normal level 80mg exforge sale. There is growing evidence that prevalence is declining, possibly due to increased use of folate supplementation. It is incompatible with life, with babies rarely living more than a few hours if they are not stillborn. Spina bifida Incomplete fusion of vertebrae potentially allowing herniation of all or part of spinal cord. They may be associated with a chromosomal abnormality, commonly trisomy 21 or 18, and with many other congenital abnormalities. Where isolated, many can be surgically corrected at birth, leading to a good quality of life. However, these features may cause considerable parental anxiety as they are often, mistakenly, seen as abnormalities. It requires ultrasound guidance and is usually performed transabdominally, occasionally transcervically. It involves aspiration of amniotic fluid which contains fetal cells shed from the skin and gut. This may be pleural or pericardial effusions, ascites, skin oedema, polyhydramnios, or placental oedema. Pathophysiology the mechanism for the development of hydrops appears to be due to an imbalance of interstitial fluid production and inadequate lymphatic return. This can result from congestive heart failure, obstructed lymphatic flow, or decreased plasma osmotic pressure. Twin-to-twin transfusion syndrome Recipient from volume overload and donor from anaemia (see b Twin-to-twin transfusion syndrome, p. In the 3rd trimester, delivery may be a better alternative than in utero treatment. If severe polyhydramnios is present, removal of excess amniotic fluid (amnioreduction) may reduce the risk of preterm labour. Consider giving steroids before the procedure as it carries a small risk of triggering preterm labour. Treatable causes of non-immune fetal hydrops Fetal anaemia In utero blood transfusion may be performed. Twin-to-twin transfusion syndrome Laser photocoagulation of placental anastomoses. These immunoglobulin (IgG) antibodies cross the placenta and cause fetal red blood cell destruction. The ensuing anaemia, if severe, precipitates fetal hydrops, which is often referred to as immune hydrops. Rhesus blood groups Consists of three linked gene pairs; one allele of each pair is dominant-C/c, D/d, and E/e. There are only five antigens (d is not an antigen; it merely implies absence of D). Antibodies may persist for weeks, causing continued haemolysis in the neonate; this requires careful monitoring with haematocrit measurements. This prevents her own immune system from recognizing them and therefore becoming sensitized. If this is suspected a Kleihauer test should be performed as the standard dose of anti-D may not be sufficient. Attaining this potential relies on such factors as a healthy mother, well-functioning placenta, and the absence of pathology. If the in utero circumstances are less than ideal, the fetus will fail to achieve its full potential growth and fetal well-being may be compromised. This method is not perfect as it includes small, but healthy babies, and average-sized unhealthy babies that should have been born big. Some of these factors have been used to generate customized growth charts which aim to identify the optimal growth curve for an individual fetus. The aim is to continue the pregnancy safely for as long as possible, thereby decreasing the problems associated with prematurity, but deliver before the fetus becomes excessively compromised. The effects appear to last into adulthood, with a stimulus or insult at a critical, sensitive period of early life having permanent effects on structure, physiology, and metabolism. People who were small or disproportionate (thin or short) at birth have been found to have higher rates of coronary heart disease, high blood pressure, high cholesterol concentrations, and abnormal glucose-insulin metabolism (Barker hypothesis). Nearly 1/100 babies in developed countries are stillborn and 1/3 of all stillbirths occur in babies that are small for dates. Any uteroplacental shortfall becomes more critical as the fetus gets bigger, and there is an extraordinary rate of growth during the last weeks of pregnancy. The only intervention is delivery so monitoring is not useful if the fetus could not survive if it were delivered, i. Ultrasound assessment of fetal growth Accurate knowledge of the age of the fetus is required. Auscultation of the fetal heart Auscultation of the fetal heart merely confirms the fetus is alive. It provides no predictive information done (with hand-held Doppler or a Pinard stethoscope) as part of a standard antenatal examination.
Purchase exforge 80 mg overnight delivery
The antigen that is so trapped persists for long periods blood pressure eating discount 80 mg exforge with mastercard, in contrast to the antigen that is captured by the medullary macrophages. One cell binds immune complexes, and the second cell, with beaded dendrites, binds to the areas of the complexes. This process constitutes what is called the alternative antigen presentation pathway,276 to distinguish it from the more conventional antigen capture and presentation. Antigen complexes are poor immunogens because they deliver a down-regulatory signal to the B cell that is related to the phosphorylation of the immune receptor Tyr-based inhibitory motif on the B-cell Fc receptor. The veils do not contain organelles, and their movement must therefore be generated at a distance. Their nucleus is highly convoluted, and the cytoplasm contains bundles of microfilaments. Only approximately 4% of veiled cells contain a Birbeck granule, but all possess a large vacuole under the cell surface. After contact sensitization, Langerhans-like cells appear in the dermal lymphatics, and an increase in the number of veiled cells in the lymph that drain the area is noted. Their function possibly consists of transporting antigens into the paracortical area, where they present them to T lymphocytes. Thus, a cellular chain for antigen transport exists in which more than one cell type takes part. The labeled cells migrated to the periarteriolar sheath of the spleen or the paracortex of the lymph node. A: Scanning electron micrograph that illustrates a follicular dendritic cell (arrow) with beaded dendrites. B: Transmission electron micrograph of a lymphocyte from the germinal center with two iccosomes in contact with its surface (arrows). A novel in vivo follicular dendritic cell-dependent iccosome-mediated mechanism for delivery of antigen to antigen-processing cells. First, they change their chemokine receptors to migrate to the T-cell areas of the draining lymph nodes. In addition to these molecular immunologic changes, morphologic signs of their activated state become apparent, with long cytoplasmic dendrites reaching out between T cells. The co-stimulatory pathway up-regulates bcl-xL and prevents Fas-mediated apoptosis of newly activated T cells. After the peak of clonal expansion, the number of antigenspecific T cells falls owing to cell death, which may occur via Fas-dependent or activation-induced cell death. In the border between the paracortex and the mantle of the follicle, they interact with B cells. B cells move inside the follicle to the pole that is adjacent to the T-cell zone (dark zone) and begin intensive proliferation. B cells undergo mutations in the V gene at a high rate (somatic hypermutation) and change their IgM to another Ig class (class switch). When proliferation subsides, centroblasts move to the center of the germinal center-i. Surviving cells have higher affinity than their predecessors (affinity maturation). Depending on the instructions that are received, some centrocytes go down memory lane, whereas others become plasma cells. Of the surviving activated T cells, most of them move out of the lymph node as memory T cells, but a small portion migrates to the edges of the paracortex next to the follicle, and B cells also move to the same location. Some of the activated B cells differentiate at the edge of the T-cell zone into plasma cells, secreting IgM and migrating to the medullary cords and on to the bone marrow. The centroblasts are in rapid cell cycle and do not increase in number, but they give rise to a progeny of centrocytes that are nondividing and express surface Ig. B cells require and receive multiple signals from different cells and molecules for survival. The decision between life or death, a process known as selection, takes place within the light zone as the B cell emerges from the dark zone. Affinity maturation is associated with somatic diversification that occurs during hypermutation. As shown by studies in mice, the D and J regions of the H chain (16 amino acids long) of the antibodies that are synthesized 7 days after immunization have no residue changes that could be attributed to mutations. However, by 14 days the antibodies carry somatic mutations that have resulted in increase of the affinity. In mice that are transgenic for Tnfsf13B, the T2 and marginal-zone compartments of B cells are enlarged. The net result is a proportional increase in the survival of the cells that traverse each stage. T-cell memory is an "operational property of the whole animal or immune system,"365 and, under this broad definition, the requirements for its maintenance and the delivery of specialized functions, although precisely regulated, depend on complex microenvironmental influences. In other words, the antibody retains the same specificity for antigen but changes all effector functions that are determined by the H chain domains, such as complement fixation and phagocytosis. It is called switch recombination, because it occurs between the switch (S) regions. Switch recombination occurs in mature B cells after exposure to antigen, T-dependent or -independent; simply by T-cell signals alone in the absence of antigen. A unique sequence 5 to each I exon determines the selectivity of the switch recombination, acting as the site for binding of a series of regulatory proteins that are expressed after activation. The I region targets class switching to specific S regions that initiate transcription,373 and an intact I region is required for recombination. The intronic enhancer (Em) makes Sm accessible for recombination with other downstream S regions ("accessibility model"). The S region of the new Ig gene is brought in juxtaposition to Sm, and the intervening sequence is looped out (B). Usually, the editing enzyme is associated with other proteins, forming an editing complex or editosome. In this respect, it differs from V(D)J recombinase, which targets consensus heptamer and nonamer sequences around the V, D, and J segments. If, for example, the class switch is between the IgM constant gene and the IgA-1 constant gene. Thus, after the IgM/IgA class switch, the IgA antibody has the same specificity as the IgM. The mutations are largely confined to the V domains395,396 and occur in the framework regions as well as the hypervariable regions. The third cluster is dominated by electrostatic interactions and stabilizes the complex. The former is expressed primarily in hematopoietic tissues, whereas the latter is expressed in several other tissues. In this respect, it plays a major role in allergic reactions and protection against helminthic infestations. The receptor subsequently phosphorylates cytoplasmic substrates that link to downstream signaling pathways. It affects the terminal differentiation of B cells and is a single polypeptide of a total of 134 amino acids with a molecular weight of 30 to 40 kDa. The membrane proximal module contains the recognition contact sites for the a chain. Binding of the cytokine induces dimerization of the receptor through free Cys that is present in the a subunit and in the first domain of the bc chain. The disulfide bonds that are formed involve the a chain of one receptor with the bc chain of the second receptor and vice versa. Because the bc chain is shared by different cytokines, there are sites that are specific for shared interactions. It is also involved in the pathology of several diseases, such as rheumatoid arthritis, proliferative glomerulonephritis, and multiple myeloma.
Exforge 80 mg visa
Effect on sport hemolysis of cold water leg immersion in athletes after training sessions heart attack racing purchase exforge 80mg online. Hemolytic anemia associated with severe hypophosphatemia in a renal transplant recipient. Traumatic haemolysis after heart valve replacement: a comparison of haematological investigations. Treatment of hemolytic anemia due to red blood cell fragmentation using recombinant human erythropoietin. Wang this chapter discusses hemoglobin (Hb) variants that cause alterations in erythrocyte morphology and rheology. Hb S, or sickle Hb, is a variant Hb of tremendous clinical importance due to its high prevalence and worldwide distribution. Long before they were recognized in the Western hemisphere, sickling disorders were known in Africa by onomatopoeic names denoting the recurrent, unrelenting, and painful nature of the crises. The term sickle cell anemia was first used in 1922, when it was recognized that a common African ancestry was present in all initial cases described. In the heterozygous state, red cells contain both normal adult Hb (Hb A) and the variant Hb. Because they rarely have phenotypic expressions of clinical significance, heterozygotes are said to have the trait for that abnormality, for example, sickle cell trait. In addition, disease may result from the combination of two variant hemoglobins or from a variant Hb and an interacting thalassemia gene. A second haplotype is prevalent in Senegal and the African West Coast, and a third haplotype is seen in the Central African Republic (Bantu-speaking Africa). The same three haplotypes are associated with the bS gene in black Americans and Jamaicans. Taking into account recent trends of decreased mortality among children, approximately 100,000 cases of sickle cell disease would be expected among African Americans in the United States. Without selective advantage to Hb S trait, the sickle gene would have been eliminated. The most widely accepted theory to account for the remarkable stability of the sickle gene in Africa is that of balanced polymorphism. This ostensibly minor change in structure is responsible for profound changes in molecular stability and solubility. Some of the sickling syndromes lack significant pathologic potential, but they are easily confused with clinically aggressive disorders on the basis of laboratory evaluation; consequently, precision in diagnosis is essential both to proper clinical management and to meaningful genetic counseling. The highest prevalence of Hb S in the world is in sub-Saharan Africa, followed by the Arabian and Indian subcontinents. It molecular basis of sickling An abundance of information indicates that the distortion of cells containing Hb S is the result of Hb polymerization. Because there are 20 surface amino acid differences between bS and g chains and only a single residue difference between bS and bA chains, it is not surprising that Hb F is excluded from Hb S polymers to a greater extent than Hb A. By measuring the minimum gelling concentration of various mixtures of hemoglobins, the extent of interaction can be quantitated. Deoxy Hb S molecules copolymerize most effectively with other Hb S molecules and, in decreasing order, with Hb C, D, O Arab, A, J, and F. The delay period between deoxygenation and polymerization is attributed to nucleation processes, in which Hb S tetramers form small aggregates without modification of internal viscosity. When these aggregates reach a critical mass, a rapid addition of free Hb units occurs to form fibers that then undergo alignment to form a tactoid. Sickling in the large veins does not produce vaso-occlusion, but the cell membrane may be damaged, resulting in a loss of water and a shorter delay time in subsequent trips through the circulation. If the delay time is less than 1 second, gelation can occur while the cell is in one of the narrow vessels of the microcirculation. Because the cell is much less deformable, it may not be able to "squeeze" through and may become transiently or permanently stuck. Under physiologic conditions, the delay between complete deoxygenation and erythrocyte sickling is 2 seconds. The delay time, however, is strongly influenced by changes in Hb concentration, the presence of hemoglobins other than Hb S, temperature, pH, and 2,3-diphosphoglycerate. Upon oxygenation, these polymers dissolve or "melt," and the sickle erythrocyte loses most of those pathologic properties caused by the presence of polymer. If the concentration of Hb S in such solutions or in the red cell approaches 30 g/dl, a semisolid gel forms. Structure of Hemoglobin S Polymer the structure of the deoxygenated Hb S polymer has been deduced from studies involving the use of electron microscopy. Physiologic Determinants of Polymerization the equilibrium of Hb S between its liquid and solid phases is determined by four variables: oxygen tension, Hb S concentration, temperature, and hemoglobins other than Hb S. Polymerization occurs only with deoxygenation, which results in a fall in oxygen affinity, thereby stabilizing the deoxy state. Part B shows the global distribution of malaria (red) before intervention to control malaria. In a study of cultured human red cells and mice, the induction of adenosine A(2B) receptor led to increased levels of 2,3-diphosphoglycerate in the cells, consequently inducing more sickling. Arterial blood, having a high oxygen saturation, contains fewer sickle cells than blood collected from various sites in the venous circulation. Predictably, red cells containing relatively more Hb F sickle less readily and survive longer than cells containing little Hb F. This phenomenon may be monitored directly with light or scanning electron microscopy. Membrane Alterations Red cell sickling is associated with reversible membrane changes. With repeated cycles of sickling and unsickling, aberrations in membrane function and structure become increasingly pronounced, culminating in fixation of the membrane in the sickled configuration. Although total cellular Ca2+ is increased, it is compartmentalized in cytoplasmic vesicles, resulting in normal levels of free cytoplasmic Ca2+ and prevention of dysfunction of the inner membrane. Reversible permeability pathways for Na+, K+, Mg2+, and Ca2+, sometimes referred to as the sickling-induced pathway, are the result of ionic shifts affecting cell hydration (58). The antimycotic agent clotrimazole is an inhibitor of the Gardos channel and prevents dehydration of sickle cells in vitro and in vivo. This phospholipid may initiate blood clotting by enhancing the conversion of prothrombin to thrombin, as suggested by the findings of increased plasma levels of fragment 1. Compared with normal red cells, sickle cells are 2 to 10 times more adherent to bovine and human endothelial cells. Furthermore, red cell deformability has a strong positive correlation with the frequency and severity of pain crises. When examined under dynamic conditions, red cell adherence is noted primarily at sites of turbulence rather than where flow is laminar. The distribution of negative charges on membranes of sickle red cells is patchy and interrupted,63 creating surface areas that may have an electrostatic attraction for other cells. Alternatively, abnormal adherence may be a derivative of cellular dehydration, which induces an abnormality of negative charge distribution in normal red cells similar to that of sickle red cells. Induction of excessive free radical generation in normal red cells is associated with increased adherence under conditions that allow the influx of calcium. B: Oxygenated irreversibly sickled cells are smooth in texture and outline but are ovoid or boatlike in shape. C: Partial deoxygenation causes the cells to assume bizarre shapes with spikes, spicules, and filaments that protrude from the cells. D: More complete deoxygenation causes the cells to assume sickled shapes with longitudinal surface striations. These conditions/factors also cause increased adhesion of sickle cells to endothelium in vitro. Thrombospondin levels are elevated in sickle cell patients during crisis,75 perhaps as a result of platelet activation. Coagulopathy might cause thrombospondin release and precipitate vaso-occlusion in microvessels, and dehydration-induced vasopressin elevation might stimulate von Willebrand factor release and precipitate vaso-occlusion in large postcapillary venules. Circulating activated endothelial cells have been assayed using immunohistochemical examination of buffy coat smears with antiendothelial cell antibodies.
