

Generic evista 60mg on-line
Most placentas invert with traction at the time of delivery womens health of augusta order evista online now, and the fetal membranes cover the maternal surface. It is important to reinvert the membranes and examine all surfaces of the placenta and membranes, looking for abnormalities. Table 5-2 lists pregnancy complications or conditions that are diagnosable at birth through examination of the placenta. B Fetal blood vessel cases of meconium aspiration syndrome, for which legal questions may arise as to whether the aspiration occurred before or during labor. If the membranes are deeply stained, the passage of meconium by the fetus may have predated onset of labor; therefore aspiration could have occurred before labor. The umbilical cord should also be examined for the number of vessels and their insertion into the placenta. Vessels on the fetal surface of the placenta should be examined for evidence of clotting or thrombosis. Besides mediating the transplacental exchange of gases and nutrients, the placenta also synthesizes glycogen with a significant turnover of lactate. Hormones secreted by the placenta have an important role for the fetus and the mother. Human placental lactogen mobilizes the breakdown of maternal fatty acid stores and ensures an increased supply of glucose to the fetus. B, Mesenchymal cells invade the cytotrophoblast 2 days after formation of the primary villi to form secondary villi. C, Blood vessels arise de novo and eventually connect with blood vessels from the embryo, forming tertiary villi. An odor may suggest infection, and cultures of the placenta may be beneficial (Benirschke and Kaufmann, 2000). Greenish discoloration may represent meconium staining or old blood; placentas with such discoloration should be sent to the pathologist for complete histologic examination. Maternal blood flows into the intervillous spaces in funnel-shaped spurts, and exchanges occur with the fetal blood as the maternal blood flows around the villi. Note that the umbilical arteries carry deoxygenated fetal blood to the placenta, and the umbilical vein carries oxygenated blood to the fetus. In addition, the cotyledons are separated from each other by decidual septa of the maternal portion of the placenta. In this drawing, only one mainstem villus is shown in each cotyledon, but the stumps of those that have been removed are shown. In addition, the placenta is a rich source of estrogen, progesterone, and glucocorticoids. Whereas progesterone maintains a quiescent, noncontractile uterus, it also has a role in protecting the conceptus from immunologic rejection by the mother. Placental transport is another important function, efficiently transferring nutrients and solutes that are essential for normal fetal growth. Endovascular transformation ensues as endovascular trophoblasts migrate into and colonize the spiral arteries, almost reaching the myometrium. This results in wide-bore, low-resistant capacitance blood vessels as observed in normal pregnancy. In contrast, shallow trophoblast invasion and incomplete transformation of spiral arteries is a common feature of preeclampsia and intrauterine growth restriction. The syncytiotrophoblast layer of the placenta is an important site of exchange between the maternal blood stream and the fetus. In addition to simple diffusion, syncytiotrophoblasts facilitate exchange by transcellular trafficking that utilizes transport proteins such as the water channels (aquaporins). In healthy women who are not pregnant, uterine blood vessels receive approximately 1% of the cardiac output to maintain the uterus. During pregnancy, these same vessels must support the rapidly growing and demanding placenta and fetus. This evolutionary challenge is addressed by remodeling of the spiral arteries, converting them into large, thin-walled, dilated vessels with reduced vascular resistance. Trophoblastorchestrated artery remodeling is an essential feature of normal human pregnancy. The precise period when trophoblast invasion of decidua and spiral arteries ceases is not clear. Although our understanding of the molecular events underlying spiral artery remodeling in pregnancy remains poor, efficient trophoblast invasion is an essential feature. The first wave is during the first trimester, when the invasion is limited to the decidual part of the spiral artery. The second wave is during the late second trimester involving deeper trophoblast invasion, reaching the inner third of myometrial segment. Displacement of the endothelial lining of spiral arteries by the invading trophoblasts further results in uncoiling and widening of the spiral artery, ensuring the free flow of blood and nutrients to meet the escalating demands of the growing fetus (Kham et al, 1999; Pijnenborg et al, 1983). A lack of spiral artery remodeling with shallow trophoblast invasion has been associated with preeclampsia. During the process of invasion in a normal pregnancy, cytotrophoblasts undergo phenotypic switching, with a loss of E-cadherin expression, and they acquire vascular endothelial-cadherin, platelet-endothelial adhesion molecule-1, vascular endothelial adhesion molecule-1, and 4 and v3 integrins (Bulla et al, 2005; Zhau et al, 1997). Although the exact gestational age at which trophoblast invasion ceases is not known, recent studies have shown that late pregnancy trophoblasts loose the ability to transform the uterine arteries. Using a novel dual-cell in vitro culture system that mimics the vascular remodeling events triggered by normal pregnancy serum, we have shown that first- and third-trimester trophoblasts respond differentially to interactive signals from endothelial cells when cultured on the extracellular matrix, matrigel. Term trophoblasts not only fail to respond to signals from endothelial cells, but they inhibit endothelial cell neovascular formation. This unique maternal and fetal cell interactive model under the pregnancy milieu offers a potential approach to study cell-cell interactions and to decipher inflammatory components in the serum samples from adverse pregnancy outcomes (Kalkunte et al, 2010). One site is the syncytiotrophoblasts covering the placental villi that are bathed in maternal blood, and the other is by the invading trophoblasts in the decidua. A representative micrograph of trophoblasts-endothelial cell interactions on matrigel is shown. Endothelial cells and trophoblasts are labeled with red and green cell tracker respectively, were independently cultured (A to E) or cocultured (F to I) on matrigel. Recently the role of specialized T lymphocytes, termed regulatory T cells (Tregs), in tolerogenic mechanisms has emerged. Tregs act by controlling the autoreactive T cells that have escaped negative selection from the thymus, and they restrain the intensity of responses by T cells reactive with alloantigens and other exogenous antigens. This unique functional capability to suppress responses to tissue-specific self-antigens that escape recognition by T cells during maturation is due to tissue specific expression and alloantigens, particularly in the epithelial surfaces where tolerance to nondangerous foreign antigen is essential to normal function. This capability enables Tregs to play a unique role at the maternal-fetal interface. Their cell numbers increase in blood, decidual tissue, and lymph nodes draining the uterus during pregnancy. Recent evidence suggests that fetal Tregs also play a vital role in suppressing fetal antimaternal immunity against maternal cells that cross the placenta (Mold et al, 2008). In the absence of Tregs the allogeneic fetus is rejected, suggesting their critical role in normal pregnancy. Unexplained infertility, spontaneous abortion, and preeclampsia are associated with proportional deficience, functional Treg deficiency, or both. Switching off or turning on genes as well as tissue-specific variation in gene expression contributes to this diversity. Gene-environment interactions resulting in epigenetic changes in the placenta during the critical window of development can influence fetal programming in utero, with predisposing health consequences later in life. Human placentation displays many similarities with tumorigenesis, including rapid mitotic cell division, migration, angiogenesis and invasion, and escape from immune surveillance. This finding suggests that a distinct pattern of tumor-associated methylation can result in a series of epigenetic silencing events necessary for normal human placental invasion and function (Novakovic et al, 2008). Moreover, the transcription factor regulating trophoblastic fusion protein syncytin, which is essential for the syncytialization of trophoblasts, is regulated by histone acetyl transferase and histone deacetylase activity (Chuang et al, 2006). Healthy development of the placenta requires efficient metabolic, immune, hormonal, and vascular adaptation by the maternal system as well as the fetus. Maternal factors such as ascending infections, obesity, hypertension, genetic predisposition such as gene polymorphism of the pregnancy-compatible cytokine milieu, and environmental exposure could also contribute to the placental pathology.
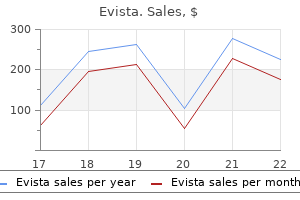
Generic 60mg evista otc
Moffett A womens health lowell general order 60 mg evista amex, Loke C: Immunology of placentation in eutherian mammals, Nat Rev Immunol 6:584-594, 2006. Normal fetal growth is determined by a number of factors, including genetic potential, the ability of the mother to provide sufficient nutrients, the ability of the placenta to transfer nutrients, and intrauterine hormones and growth factors. The pattern of normal fetal growth involves rapid increases in fetal weight, length, and head circumference during the last half of gestation. During the last trimester, the human fetus accumulates significant amounts of lipid. The birthweight for gestational measurements among populations has been shown to increase over time; therefore, standards for normal fetal growth require periodic reevaluation for clinical relevance. These increases in birthweight for gestational age over time are attributed to improvements in living conditions and maternal nutrition and changes in obstetric management. Variations in fetal growth have been identified in diverse populations and are associated with geographic locations (sea level versus high altitude), populations (white, African American, Latino), maternal constitutional factors, parity, maternal nutrition, fetal gender, and multiple gestations. In this chapter, we discuss these factors in greater detail and critically review the long-term effects of abnormal fetal growth. The duration of pregnancy has become an integral component of prenatal growth assessment, and all currently prevailing definitions of fetal growth are specific for gestational age. Any error in dating will lead to misclassification of the infant, which can have significant clinical implications. For example, some nomograms are based on approximating the gestational age to the nearest week, whereas others use the completed weeks. The birthweight charts are also affected by other variables that may limit their reliability. Many of these variables, such as fetal gender, race, parity, birth order, parental size, and altitude, contribute to the normal biologic variations in human fetal growth. There is continuing controversy regarding whether the reference growth charts should be customized by multiple variables or developed from the whole population. The customized approach predicts the optimal growth in an individual pregnancy and therefore specifically defines suboptimal growth for that pregnancy. In recognition of the utility of a national standard, a population-based reference chart for fetal growth has been developed from all the singleton births (more than 3 million) in the United States in 1991 (Alexander, 1996). More recently, a similar national population-based fetal growth chart, which is also sex specific, has been developed in Canada (Kramer et al, 2001). There is insufficient evidence about whether one approach is superior to the other in improving the perinatal outcome. Various definitions appear in the medical literature, making comparisons between studies difficult. In addition, investigators have shown that the prevalence of fetal growth restriction varies according to the fetal growth curve used (Alexander et al, 1996). However, these definitions do not make a distinction among infants who are constitutionally small, growth restricted and small, and not small but growth restricted relative to their potential. As an example, as many as 70% of fetuses with a weight less than the 10th percentile for gestational age at birth are small simply because of constitutional factors such as female sex or maternal ethnicity, parity, or body mass index; they are not at high risk of perinatal mortality or morbidity. In contrast, true fetal growth restriction is associated with numerous perinatal morbidities. This has clinical relevance to perinatologists and neonatologists, because many of the tiniest premature neonates in the neonatal intensive care units are probably growth restricted. McIntire et al (1999) reported a threshold of increased adverse outcomes in infants born with measurements less than the 3rd percentile and suggested that this level of restriction represents a clinically relevant measurement. Other researchers have found higher rates of neonatal complications when the 15th percentile of birthweight is used as a cutoff level (Seeds and Peng, 1998). Even when a normal intrauterine growth pattern is established for a population, somewhat arbitrary criteria are used to define growth restriction. Weight (and then length) of infants with asymmetric growth retardation is affected, with a relatively normal or "headsparing" growth pattern. Asymmetric patterns generally develop during the third trimester, a period of rapid fetal growth. However, now that fetal surveillance is more common, asymmetric growth restriction is often diagnosed in the second trimester. Factors that are well recognized to limit the growth of both the fetal brain and body include chromosomal anomalies. In a simplified manner, these factors can be grouped as fetal, placental, or maternal in origin. Fetal Causes of Growth Restriction Fetal factors affecting growth include gender, familial genetic inheritance, chromosomal abnormalities, and dysmorphic syndromes. Other studies have similarly found that fetal growth restriction is more common among infants with malformations. Fetal gender also influences size, with male infants showing greater intrauterine growth than female infants (Glinianaia et al, 2000; Skjaerven et al, 2000; Thomas et al, 2000). Etiologies of Fetal Growth Restriction the epidemiology of fetal growth restriction varies internationally (Keirse, 2000). In developed countries, the most frequently identified cause of growth restriction is smoking, whereas in developing countries, maternal nutritional Placental Causes of Growth Restriction In mammals, the major determinant of intrauterine growth is the placental supply of nutrients to the fetus (Fowden et al, 2006). In many species, fetal weight near term is positively correlated to placental weight, as a proxy measure of the surface area for maternal-fetal transport of nutrients. Fetal weight near term is positively correlated to placental weight, and the nutrient transfer capacity of the placenta depends on its size, morphology, blood flow, and transporter abundance (Fowden et al, 2006). In addition, placental synthesis and metabolism of key nutrients and hormones influences the rate of fetal growth (Fowden and Forhead, 2004). Changes in any of these placental factors can, therefore, affect intrauterine growth; however, the fetus is not just a passive recipient of nutrients from the placenta. The fetal genome exerts a significant acquisitive drive for maternal nutrients through adaptations in the placenta, particularly when the potential for fetal and placental growth is compromised. Placental maturation at the end of pregnancy is associated with an increase in substrate transfer, a slowing (but not cessation) of placental growth, and a plateau in fetal growth near term (Fox, 1997). Abnormalities of placental growth, senescence, and infarction have been shown to affect fetal growth. The placentas from pregnancies complicated by poor fetal growth have a higher incidence of vascular damage and abnormalities (Pardi et al, 1997). Placental growth in the second trimester correlates with placental weight and function and, thus, with weight at birth (Godfrey et al, 1996). Clinical conditions associated with reduced placental size (and subsequent reduced fetal weight) include maternal vascular disease, uterine anomalies (uterine fibroids, abnormal uterine anatomy), placental infarctions, unusual cord insertions, and abnormalities of placentation. Multiple gestations are associated with greater risk for fetal growth restriction. The higher risk stems from crowding and from abnormalities with placentation, vascular communications, and umbilical cord insertions. Divergence in fetal growth appears from approximately 30 to 32 weeks in twin gestation compared with singleton pregnancies (Alexander et al, 1996; Glinianaia et al, 2000; Skjaerven et al, 2000). Larger effects on fetal growth are seen with increasing number of fetal multiples. Abnormalities in placentation are also more common with multiple gestations (Benirschke, 1995). Monochorionic twins can share placental vascular communication (twin-twin transfusion), leading to fetal growth restriction during gestation. Fetal competition for placental transfer of nutrients raises the incidence of growth restriction and discordance in growth between fetuses. The rate of birthweights less than the 5th percentile is higher in monochorionic twins. Placental growth is restricted in utero because of limitation in space, leading to a higher incidence of placenta previa in multiple-gestation pregnancies. In addition, abnormalities in cord insertions (marginal and velamentous cord insertions) and occurrence of a single umbilical artery are more frequently found in multiple gestations. The higher incidence of growth restriction in multiple-gestation pregnancies is strongly associated with monochorionic gestations, the presence of vascular anastomoses, and discordant fetal growth (Hollier et al, 1999; Sonntag et al, 1996; Victoria et al, 2001). Placentas of smaller fetuses with discordant growth are significantly smaller than those of their larger twin counterparts (Victoria et al, 2001). Investigators have shown an effect of altitude on fetal growth, with infants born at high altitudes having lower birthweights (Galan et al, 2001). In these high-altitude pregnancies, the abdominal circumference is most affected (Krampl et al, 2000).
Diseases
- Tucker syndrome
- Pancreatic carcinoma, familial
- Trichodermodysplasia dental alterations
- Cholestasis
- Retrolental fibroplasia
- Hydrocephalus obesity hypogonadism
Discount evista 60 mg online
Discontinuation of protein and the provision of intravenous fluids with high caloric content are the first steps to take women's health center colonial park effective 60 mg evista. L-Arginine or L-citrulline, as well as the "scavenger drugs" sodium phenylbutyrate and sodium benzoate, may be administered. Hemodialysis might be required to control the neurotoxic hyperammonemia, which can lead to irreversible brain damage, coma, and death. It is hoped that with early identification through newborn screening, patients with urea cycle disorders will be protected by presymptomatic therapy in the neonatal period. Arginase deficiency can also present acutely with hyperammonemia as described earlier, although more frequently it manifests as developmental delay and spastic diplegia in childhood with a milder degree of hyperammonemia (Crombez and Cederbaum, 2005). However, the preanalytic processing required for succinylacetone is more involved than that required for the amino acids and acylcarnitines. These programs may rely on elevations of tyrosine for identification of this disorder. Unfortunately, moderate elevations of tyrosine that are transient occur frequently in neonates, especially those who have low birthweights and are sick, necessitating frequent requests for repeated screening with virtually no detection of tyrosinemia type I. Unfortunately moderate transient elevations of tyrosine occur frequently in neonates, especially those who have low birthweights and are sick, necessitating frequent requests for repeated screening. Virtually no cases of tyrosinemia type I have been detected based on elevated tyrosine because almost all infants with tyrosinemia type I have had normal tyrosine levels when screened (Frazier et al, 2006). Consequently, the newborn detection of tyrosinemia type I by a tyrosine marker alone is ineffective. Tyrosinemia type I leads to liver and renal tubular disease and can later result in hepatocellular carcinoma. Medium-Chain Acyl-CoA Dehydrogenase Deficiency and Other Fatty Acid Oxidation Disorders the fatty acid oxidation disorders include those in which the long-chain fatty acids cannot traverse the mitochondrial membranes to be oxidized within the mitochondrial matrix. Fatty acid oxidation is essential to supply energy as adenosine triphosphate via the Krebs cycle and as ketones in the presence of a low supply of glucose. The disorders involving defective transport concern carnitine, whereas those with defective oxidation are named according to the enzyme that is deficient (see Table 27-1). The clinical consequence of these disorders is fasting intolerance resulting in hypoketotic hypoglycemia, lethargy, hyperammonemia, metabolic acidosis, hepatomegaly, and sometimes sudden death. Tragically, before newborn screening was available, this disorder was often diagnosed only retrospectively after a sudden unexplained death, usually when postmortem examination revealed a fatty liver. The treatment for fatty acid oxidation disorders is avoidance of fasting with high-carbohydrate, low-fat feedings and, of critical importance, prompt attention to acute illnesses in which vomiting occurs. Any infant with a fatty acid oxidation disorder should be evaluated at a metabolic center. Organic Acid Disorders Organic acid disorders are a heterogenous group of disorders with a combined frequency of approximately 1 in 50,000 (Zytkovicz et al, 2001). The marker for this disease group, as for the fatty acid oxidation disorders, is an abnormal acylcarnitine pattern. If a screening result suggests an organic acidemia, a metabolic specialist should be consulted immediately. The major organic acid disorders identified in newborn screening are propionic acidemia, the methylmalonic acidemias, and isovaleric acidemia. The organic acidemias can manifest in the neonatal period with a life-threatening, sepsis-like picture of feeding difficulties, lethargy, vomiting, and seizures. Metabolic acidosis virtually always accompanies this presentation, and hyperammonemia is common. In this situation, protein administration should be discontinued and replaced by administration of intravenous fluids with high caloric content and carnitine. The effects of early diagnosis and treatment on the clinical and neurologic development of individuals affected by an organic acid disorder are currently under investigation (Albers et al, 2001b). The enzyme assay identifies only galactosemia, whereas the metabolite assay also identifies other galactose metabolic disorders, such as deficiencies of galactokinase and epimerase. Severe neonatal liver disease and portosystemic shunting caused by anomalies in the portal system can also increase the galactose level. The most rapid confirmatory test for a positive result in galactosemia screening is urine testing for reducing substance. In almost all cases of severe galactosemia, this test produces a strongly positive reaction. If the urine contains reducing substance and the infant has clinical signs of galactosemia. Approximately 70% of the patients with galactosemia carry the Q188R mutation (Elsas and Lai, 1998). Nevertheless, urine-reducing substance may be absent in infants with clinically significant variants of galactosemia. Consequently, follow-up testing should be performed for all infants with an initial positive galactosemia screening result. Biotinidase Deficiency Biotin recycling is necessary for the maintenance of sufficient intracellular biotin to activate carboxylase enzymes. Lack of biotinidase activity results in reduced carboxylase activities and an organic acid disorder known as multiple carboxylase deficiency (Wolf and Heard, 1991). The clinical features of the disorder are developmental delay, seizures, hearing loss, alopecia, and dermatitis. Initiating biotin therapy in early infancy, when the disorder is presymptomatic, seems to prevent all the features of biotinidase deficiency. For this reason, a screening test has been developed and added to newborn screening in a number of newborn screening programs throughout the world (Hart et al, 1992). The frequency of identified newborns in these programs has a wide range, from 1:30,000 to 1:235,000. Almost all identified infants have been asymptomatic and have remained normal with biotin treatment. Galactosemia Galactosemia typically manifests in the neonatal period as failure to thrive, vomiting, and liver disease (Hughes et al, 2009). Death from bacterial sepsis, usually caused by Escherichia coli, occurs in a high percentage of untreated neonates (Levy et al, 1977). Some screening programs use a metabolite assay for total galactose (galactose and galactose-1-phosphate) to detect galactosemia. The major clinical features of untreated congenital hypothyroidism are growth retardation and delayed cognitive development leading to mental deficiency. If treatment with pharmacologic doses of T4 is initiated early, growth and mental development are normal. This situation may be due to a lack of the identifying marker abnormality at the time of specimen collection. Specifically, the T4 level during the first 24 hours of life in an affected infant might not yet be sufficiently decreased for identification because of persistence of maternally transmitted T4. The reported false-positive rates of screening for congenital hypothyroidism range from approximately 0. To avoid missing congenital hypothyroidism, screening programs require a second blood specimen from each of these infants. Infants with a positive screening test result should not be labeled as having congenital hypothyroidism until diagnostic testing confirms the disorder. If congenital hypothyroidism is confirmed, however, administration of T4 should be started without delay to prevent irreversible brain damage. Cross-reacting steroids are produced by residual fetal adrenal cortex or result from decreased metabolic clearance by an immature liver. If the infant shows signs of illness or has ambiguous genitalia, serum electrolytes should be measured. If these results indicate hyponatremia and hyperkalemia, the infant should be hospitalized without delay, and the electrolyte imbalance should be corrected immediately. The major goal of this testing is to identify infants with sickle cell disease so that they can be given penicillin prophylaxis to prevent pneumococcal septicemia. Additional benefits of early detection are early referral to a comprehensive sickle cell program and early education and genetic counseling for parents (Smith and Kinney, 1993).
Buy evista 60mg without a prescription
Outlet forceps-the fetal vertex is visible at the labia without manually separating them breast cancer bows buy evista 60mg without prescription, and the fetal skull has reached the pelvic floor 2. Low forceps-the leading point of the fetal skull is greater than 2 cm beyond the ischial spines 3. Mid forceps-the fetal head is engaged and is beyond the level of the ischial spines; the forceps should be applied only if cesarean delivery is not quickly or imminently possible, and the fetus is in distress; or there should be a high likelihood that the forceps operation will be successful 4. The forceps are further divided into whether they are rotational (sagittal suture is >45 degrees from the midline) or nonrotational (sagittal suture <45 degrees from the midline). Maternal exhaustion or inability to push (endotracheal intubation with sedation or paralysis; neuromuscular disease) 2. Maternal contraindications to pushing (cardiopulmonary disease, cerebrovascular aneurysm) 4. Today, with the widespread availability of cesarean delivery, considerations turn to providing the best neonatal outcome possible; therefore the difficult forceps deliveries of the past have been abandoned. Nevertheless, forceps still play a crucial role in modern obstetrics, and if judiciously used can provide a safer alternative to cesarean delivery for both mother and baby. Furthermore, residency training in operative vaginal delivery has dramatically decreased over the past 30 years, potentially increasing fetal risk (Benedetto et al, 2007). The incidence of operative vaginal delivery in the United States is approximately 5% (1 in 20 deliveries), ranging from 1% to 23% with a 99% success rate (Menacker and Martin, 2008). The prevalence of forceps use varies widely by region (highest in the South), and recent estimates show that it accounts for approximately 25% (1:4 ratio with vacuum extractions) of all operative vaginal deliveries (Benedetto et al, 2006). Unfortunately, the prevalence of low, outlet, and mid-forceps (the application of forceps when the head is engaged but the leading point of the skull is more than +2 cm station) deliveries nationally is not known, nor is the rate of rotational and nonrotational forceps use. Moreover the indication for forceps use varies widely by clinical situation, and the neonatal morbidity that can result from a "difficult pull" in a patient with a transverse arrest with marked fetal asynclitism may be different from the quick delivery of a 2600-g fetus whose mother is unable to push, even if both deliveries are by low-forceps. Nevertheless, what large studies show that long-term and short-term neonatal morbidity from outlet or lowforceps delivery is uncommon In 2009, Prapas et al (2009) noted that the rate of vacuum- versus forceps-assisted deliveries had increased and that different maternal and neonatal outcomes have been proposed. The aim of their study was to compare the short-term maternal and neonatal outcomes between vacuum and forceps delivery. They conducted a medical record review of live born singleton, vacuum- and forceps-assisted deliveries. Of 7098 deliveries, 374 were instrument assisted, 324 were conducted by vacuum (86. The incidence of third-degree lacerations and periurethral hematomas was similar between vacuum and forceps (3. The rate of neonates with Apgar scores 6 at 1 min was significantly higher after forceps compared with vacuum delivery (18% vs. The rate of neonatal trauma and respiratory distress syndrome did not differ significantly between the two groups. The conclusion was that both modes of instrumental vaginal delivery are safe in regard to maternal morbidity and neonatal trauma (Prapas et al, 2009). Alternatively, Benedetto et al, (2007) found that in healthy women with antenatally normal singleton pregnancies at term, instrument-assisted deliveries are associated with the highest rate of short-term maternal and neonatal complications. Of the 332 women who underwent an operative vaginal delivery, 201 met study criteria and were analyzed, with 54% forceps-assisted deliveries. There are few randomized prospective trials specifically addressing the issue of neonatal morbidity arising from forceps operations, yet those that exist suggest no significantly increased risk from operative vaginal delivery when compared with spontaneous birth. Yancey et al (1991) randomized 364 full-term women at +2 station to elective outlet forceps delivery or unassisted birth. Neonates were examined at birth and 72 hours of age, and cranial ultrasound examinations were performed by neonatologists 24 to 72 hours after birth. Several retrospective, population-based studies have further examined the issue of potential adverse neonatal sequelae arising from forceps procedures. They found that mid-forceps operations compared with a cesarean section from a comparable station resulted in greater neonatal resuscitation requirements, lower umbilical artery pH, and birth trauma, defined as nerve injuries or fractures. There was no increased risk of trauma from low forceps procedures compared with cesarean delivery, although the prevalence of arterial cord pH less than 7. It must be noted, however, that this study was not randomized and that an abdominal delivery group is not necessarily an appropriate control for operative vaginal delivery. Nevertheless, this large population-based study (from a database of 20,831) emphasizes the relative safety of outlet and low-forceps procedures, casting some doubt as to the safety of mid-forceps operations. The role of mid-forceps delivery in modern obstetrics, specifically in regard to rotation of 45 degrees or greater, has incited much controversy. The difficulty lies in the fact that no randomized trials exist comparing mid-forceps operations and other modes of delivery, and it is unlikely that any will be done in the near future given that training in this type of delivery has declined among obstetric residents in the United States (Kozak and Weeks, 2002; Learman, 1998). Hankins et al reported a case-control study of 113 rotational forceps of 90 degrees or greater matched to 167 controls with rotation 45 degrees or less. They found no significant differences in major injuries, defined as skull fracture, brachial plexus, facial or sixth nerve palsy, and subdural hemorrhage. There were also no differences in the prevalence of cephalhematoma, clavicular fracture, or superficial laceration. All deliveries were performed with Kielland forceps by resident staff members, with attending supervision as required. The prevalence of nerve injury in these forceps-assisted operations ranged from 2% to 3%; there were no skull fractures (Lipitz et al, 1989). The available literature supports the fact that neonatal morbidity from outlet or low forceps is exceedingly low, and comparable to spontaneous vaginal delivery. This finding comes from prospective data and large, population-based investigations. In well-trained hands, the benefit of appropriately selected candidates for mid-forceps or rotational forceps may be justifiable, although the number of physicians in the United States who are facile and comfortable with attempting these procedures is steadily declining. However, it did not gain widespread use until the 1950s, when it was popularized in a series of studies by the Swedish obstetrician Dr. By the 1970s, the vacuum extractor had almost completely replaced forceps for assisted vaginal deliveries in most northern European countries, but its popularity in many English-speaking countries, including the United States and the United Kingdom, was limited. By 1992, however, the number of vacuum-assisted deliveries surpassed the number of forceps-assisted deliveries in the United States, and by the year 2000 approximately 66% of operative vaginal deliveries were by vacuum (Ali and Norwitz, 2009; Hillier and Johanson, 1994). The situations that indicate the use of the vacuum and the requirements that must be fulfilled for its correct use are identical to those for the obstetric forceps. The device consists of a metal or plastic cup (flexible or semirigid) that is applied to the fetal vertex. Care is taken in its application to ensure that an adequate seal has been created, and that no maternal soft tissue is trapped between the suction device and the fetus. Traction is then applied to the fetal head in the line of the birth canal in an effort to assist delivery. It is generally advised that no more than three detachments occur before attempts at vacuum extraction are abandoned (Ali and Norwitz, 2009). In a laboratory experiment, Duchon et al (1988) compared the maximum force at suggested vacuum pressures (550-600 mm Hg) prior to detachments for different types of vacuum devices. They found that the average force of traction exerted before detachments ranged from 18 to 20 kg (Benedetto et al, 2007). This result is interesting to bear in mind when one considers the older data of Wylie who estimated the average tractive force required for delivery of infants weighing 15. Because of technical problems and lack of experience with this instrument, vacuum devices did not gain popularity in the United States until the introduction of the disposable cups in the 1980s. The soft cup is a pliable, funnel- or bellshaped cup, which is the most common type used in the United States. The rigid cup is a firm mushroom-shaped cup (M cup) similar to the original metal disc-shaped cup and is available in three sizes. For example, the risk of scalp laceration with the rigid Kiwi OmniCup (Clinical Innovations, Murray, Utah) was reported to be 14. These and other authors concluded that handheld soft bell cups should be considered for more straightforward occiput-anterior deliveries, and that rigid M cups should be reserved for more complicated deliveries, such as those involving larger infants, significant caput succedaneum (scalp edema), occiput-posterior presentation, or asynclitism. The vacuum extractor is widely used in the United States, but is not free of preventing neonatal injury.

Order evista 60 mg with visa
Seronegative women are therefore at risk for acquiring primary infections during pregnancy pregnancy effacement order evista 60 mg online. Primary infections pose an increased risk of transmission to the fetus, and possibly a higher risk of sequelae. Inclusion-bearing epithelial cells have been described in the semicircular canals, vestibular membrane, cochlea, and other structures of the ear. In addition, temporal bone anomalies with cochlear, vestibular, and auditory canal defects have been noted in association with hearing loss in affected infants (Davis, 1969; Myers and Stool, 1968). Findings include intrauterine growth restriction, microcephaly, ventriculomegaly, periventricular calcifications, echogenic bowel, polyhydramnios, pleural effusion, pericardial effusion, hepatosplenomegaly, intrahepatic calcifications, pseudomeconium ileus, and placental enlargement (Guerra et al, 2008; Nelson and Demmler, 1997). Infection in the symptomatic infant can involve any organ and manifests along a spectrum from mild illness to severe disseminated multiorgan system disease. Clinical features include jaundice, hepatosplenomegaly, lethargy, respiratory distress, seizures, and petechial rash. Infants with symptomatic disease are often premature and small for gestational age. A wide spectrum of disease can be observed, including hemolysis, bone marrow suppression, hepatitis, pneumonitis, enteritis, and nephritis. Common laboratory abnormalities include thrombocytopenia, anemia, abnormal levels of liver enzymes (particularly elevated transaminases), and elevated conjugated bilirubin levels. These pathologies include meningoencephalitis, calcifications, microcephaly, neuronal migration disturbances, germinal matrix cysts, ventriculomegaly, and cerebellar hypoplasia (Cheeran et al, 2009). Long-term neurodevelopmental disabilities are observed in 50% to 90% of children who are symptomatic at birth. In contrast, long-term neurodevelopment injury is strikingly less likely in congenitally infected infants who are asymptomatic at birth: when it does occur, it is typically limited to hearing deficits. Among symptomatic congenitally infected infants, longterm sequelae can include microcephaly, hearing loss, motor deficits (paresis or paralysis), cerebral palsy, mental retardation, seizures, ocular abnormalities (chorioretinitis, optic atrophy), and learning disabilities (Cheeran et al, 2009; Sharon and Schleiss, 2007). C and D, T1 flash axial (C) and sagittal (D) images of a symptomatic, congenitally infected infant demonstrating ventriculomegaly, polymicrogyria, and porencephalic cyst (arrow). Delayed-onset hearing loss usually occurs before 4 years of age (Dahle et al, 2000; Fowler et al, 1997; Rivera et al, 2002; Williamson et al, 1992), but has been reported to evolve and progress through 6 years of age and beyond. A retrospective review of symptomatic congenitally infected infants identified neurologic and radiologic sequelae in 81% of affected patients. There was a significant correlation between the severity of the initial pure-tone audiometry and the development of progressive hearing loss, in addition to a significant correlation between a less severe final pure-tone audiometry and the presence of cerebral palsy (Madden et al, 2005). The timing of these samples is important because subsequent viral isolation may represent neonatal infection acquired in the birth canal or after exposure to breast milk (Schleiss, 2006b). This "shell vial" assay has a high sensitivity and specificity, and it allows the confirmation of diagnosis within 24 hours of inoculation (Gleaves et al, 1984; Rabella and Drew, 1990). The magnitude of the systemic viral load in the congenitally infected infant can be a predictor of neurodevelopmental prognosis. Ultrasound, because of its convenience, is an appropriate initial study and is particularly valuable and sensitive in detecting periventricular calcifications and lenticulostriate vasculopathy associated with mild to moderate ventricular dilatation. Follow-up evaluation should include a multidisciplinary team approach involving a pediatric infectious diseases specialist, pediatric otolaryngologist, and child behavioraldevelopmental specialist, in addition to a physical therapist, ophthalmologist, and neurologist as needed. Although almost two thirds of the treated infants had neutropenia, it was reversible when antiviral therapy was halted. All infants with documented congenital infection should have an ophthalmologic evaluation. If chorioretinitis is present, it should be managed in consultation with an ophthalmologist and infectious diseases expert. Other investigational studies including the assessment of the therapeutic potential in infants of the prodrug of ganciclovir, valganciclovir, are needed. Ganciclovir has demonstrated teratogenic risk in some studies (Schleiss and McVoy, 2004); although this has never been demonstrated in humans, it has limited research in this area. A case report of the use of oral ganciclovir in a pregnant liver transplant patient did not show any evidence of teratogenicity (Pescovitz, 1999). Of these potential mechanisms, the most common is via breast milk (Schleiss, 2006c), with transmission in the birth canal occurring less commonly. Approximately half of these infants were ill and exhibited symptoms such as hepatopathy, neutropenia, thrombocytopenia, and sepsislike deterioration. Of these methods, freezing is the most studied and most likely to maintain the salutary immunologic properties of breast milk. When symptoms occur, they are nonspecific and vague, often described as a flulike syndrome. Potential manifestations include fever, fatigue, headache, myalgia, lymphadenitis, and pharyngitis, but these are the exception and not the rule. For several years after discovery, its role in disease was unclear, but it is now known to be the major etiologic agent of roseola infantum (exanthem subitum) and has been implicated in other clinical syndromes. Whether intrapartum transmission of these viruses can occur in the birth canal during delivery remains unknown. Perinatal transmission via this mechanism has been postulated (Joshi et al, 2000), but has not been demonstrated. Indeed, the routes of acquisition of infection and mechanisms responsible for person-to-person transmission remain uncertain. However, more recent evidence suggests that other routes of infection exist, including transmission by saliva (Pica and Volpi, 2007). There appears to be considerable regional variation in prevalence in the United States. In a population of children in south Texas, the seroprevalence was 26%, strongly suggesting that nonsexual modes of transmission predominate (Baillargeon et al, 2002). In Sub-Saharan Africa, prevalence in children is even higher, approaching 60% in some studies (Sarmati, 2004). The rash first appeared on the face and gradually spread to the trunk, arms, and legs. It initially consisted of discrete red macules that blanched with pressure and eventually became papular. An upper respiratory tract infection appeared as a secondary symptom in most children, and a lower respiratory tract infection appeared as a secondary symptom in one third of symptomatic children. Additional information on the epidemiology and modes of transmission of this pathogen, particularly in the prenatal and intrapartum period, is needed. In another study, placentas and some fetuses were studied in five cases of pregnancy interruption caused by maternal infectious mononucleosis in early gestation (Ornoy et al, 1982). Decidual lesions, consisting of perivasculitis and necrotizing deciduitis, were noted, and endovasculitis, perivasculitis, and occasional vascular obliteration were found in villi, as well as mononuclear and plasma cell infiltrates. The virus was identified in 1975 (Cossart et al, 1975) and was first linked to a disease in 1981-aplastic crisis in children with sickle cell anemia (Pattison et al, 1981). Primary infection with parvovirus B19 is commonly known as fifth disease or erythema infectiosum; it is classically described as a childhood exanthem with a "slapped cheek" appearance (Anderson et al, 1984). Considerable interest in the role of this virus in hydrops fetalis (nonimmune) and fetal aplastic crisis has evolved since the first cases of fetal death associated with maternal parvovirus B19 infection were reported in the 1980s (Brown et al, 1984; Kinney et al, 1988). A significant proportion of childbearing women are thus susceptible to infection (Markenson and Yancey, 1998; Yaegashi et al, 1998). Preconception seroprevalence to parvovirus B19 ranges from 24% to 84% (Ergaz and Ornoy, 2006). During pregnancy the risk of acquiring parvovirus B19 infection is low, ranging from 0% to 16. The risk of primary maternal infection is higher during epidemics, with reported seroconversion rates ranging between 3% (Kerr et al, 1994) and 34% (Woernle et al, 1987). It is estimated that one fourth to half of maternal parvovirus infections result in transmission of infection to the fetus (Alger, 1997; Gratacos et al, 1995; Koch et al, 1998). The vast majority of pregnancies are unaffected (Berry et al, 1992; Sheikh et al, 1992). There are conflicting reports regarding the prognosis once fetal infection has been established. A longitudinal study of fetal morbidity and mortality in more than 1000 women with primary parvovirus B19 infection in pregnancy demonstrated a risk of fetal hydrops of 3.
Syndromes
- Seeing or hearing things that are not really there (hallucinations)
- Fluid around the lung (pleural effusion)
- Acute pancreatitis
- Blue-tinged or very pale whites of eyes
- Prolonged bleeding
- Medications to treat depression (antidepressant drugs)
- Fluids by IV
Purchase evista once a day
This type of late metabolic acidosis is rarely seen menstruation hormones best order for evista, probably because of the use of special premature infant formulas and changes in regular formulas with decreased casein-to-whey ratios and lower fixed acid loads. The presence of metabolic acidosis in the newborn should be suggested by the clinical presentation and the history of predisposing conditions, including perinatal depression, respiratory distress, blood or volume loss, sepsis, and congenital heart disease associated with poor systemic perfusion or cyanosis. The cause of metabolic acidosis is often readily discernible from the history and physical examination. Specific laboratory evaluation of electrolytes, renal function, lactate, and serum and urine amino acids may be undertaken, depending on the diagnosis that is suggested clinically. It is important to remember that infants might not manifest an increased anion gap in the setting of lactic acidosis and thus require the direct measurement of lactate when a lactic acidosis is suspected (Lorenz et al, 1999). The morbidity and mortality of metabolic acidosis depend on the underlying pathologic process, the severity of the acidosis, and the responsiveness of the process to clinical management. By far, the most important intervention for an infant with a metabolic acidosis is to identify the pathologic process contributing to the acidosis and to take measures to correct it. The administration of base, such as sodium bicarbonate, as supportive therapy for metabolic acidosis is unproven in its efficacy (Aschner and Poland, 2008). Infants with lactic acidosis may not have an increased anion gap and lactate should be measured directly if suspected by the history and physical examination. The use of sodium bicarbonate in this study was associated with an increase in myocardial contractility and a reduction in afterload, albeit transient. However, there is concern for harm associated with the administration of base, including increased mortality and intraventricular hemorrhage (Papile et al, 1978; Simmons et al, 1974; Usher, 1967), increased cerebral blood volume regardless of rate of administration (van Alfen-van der Velden et al, 2006), and decreased intracellular pH with cellular injury (Lipshultz et al, 2003). If therapy with base is warranted, the clinician has three options: sodium bicarbonate, sodium (or potassium) acetate, and tromethamine. Sodium bicarbonate is the most widely used buffer in the treatment of metabolic acidosis in the neonatal period. Bicarbonate should not be given if ventilation is inadequate, because its administration results in an increase in Paco2 with no improvement in pH and an increase in intracellular acidosis. Therefore sodium bicarbonate should be administered slowly and in diluted form only to newborns with documented metabolic acidosis and adequate alveolar ventilation. Subsequent doses of sodium bicarbonate are then based on the results of additional blood gas measurements. When clinicians are faced with a chronic metabolic acidosis caused by a prematurity-related proximal renal tubular acidosis with bicarbonate wasting, many choose to replace these losses over time. In this instance, either sodium or potassium acetate can be used as an alternative to sodium bicarbonate. It has been shown in one study to be an effective alternative to sodium bicarbonate in correcting this type of acid-base abnormality when added to parenteral nutrition (Peters et al, 1997). Infants randomized to acetate had an increased base excess and pH and increased Pco2, and they received less bicarbonate boluses compared with control infants. In certain clinical situations, tromethamine can be used as an alternative buffer to sodium bicarbonate. The theoretical advantages of tromethamine over sodium bicarbonate in the treatment of metabolic acidosis of the newborn include its more rapid intracellular buffering capability, its ability to lower Paco2 levels directly, and the lack of an increase in the sodium load (Schneiderman et al, 1993). Although there are controversies regarding the actual bicarbonate space in humans, the 30% of total body weight in the formula represents its estimated volume of distribution in the neonate. Because the end-product (chelated tromethamine) is a cation that is excreted by the kidneys, oliguria is a relative contraindication to the repeated use of this buffer. Tromethamine administration also has been associated with the development of acute respiratory depression, most likely secondary to an abrupt decrease in Paco2 levels as well as from rapid intracellular correction of acidosis in the cells of the respiratory center (Robertson, 1970). Furthermore, because hypocapnia is associated with decreases in brain blood flow and a higher incidence of white matter damage, especially in the immature preterm neonate, close monitoring of Paco2 is of paramount importance when tromethamine is being used. Finally, when large doses of tromethamine are administered, hyponatremia (Seri et al, 1998b), hypoglycemia, hyperkalemia, an increase in hemoglobin oxygen affinity, and diuresis followed by oliguria can occur. Because the tromethamine solution is hyperosmolar, and because rapid infusion of tromethamine can also lower blood pressure and intracranial pressure (Duthie et al, 1994), slow infusion is recommended. Because potassium moves from the intracellular to the extracellular space in exchange for H+ when acidosis occurs, the presence of a total body potassium deficit might not be appreciated during metabolic acidosis. Hypokalemia may become evident only as the pH increases and potassium returns to the intracellular space. Furthermore, intracellular acidosis cannot be completely corrected until the potassium stores are restored. Therefore close monitoring of serum electrolytes and potassium supplementation are important during the correction of metabolic acidosis in the sick newborn. Respiratory Acidosis Respiratory acidosis occurs when a primary increase in Paco2 develops secondary to impairments in alveolar ventilation that result in an arterial pH of less than 7. Primary respiratory acidosis is a common problem in newborns, and causes include hyaline membrane disease, pneumonia owing to infection or aspiration, patent ductus arteriosus with pulmonary edema, chronic lung disease, pleural effusion, pneumothorax, and pulmonary hypoplasia. Management of respiratory acidosis is directed toward improving alveolar ventilation and treating the underlying disorder. For sick newborns, adequate ventilation must often be provided by mechanical ventilation. Therefore tromethamine should be used only as a temporizing measure in severe respiratory acidosis until alveolar ventilation can be improved. The most common causes of this type of metabolic alkalosis in the newborn period are continuous nasogastric aspiration, persistent vomiting, and diuretic treatment. Less common causes of H+ losses are congenital chloride-wasting diarrhea, certain forms of congenital adrenal hyperplasia, hyperaldosteronism, posthypercapnia, and Bartter syndrome. In the past, a metabolic alkalosis was intentionally created when sodium bicarbonate or tromethamine was used to maintain an alkaline pH to decrease pulmonary vasoreactivity in infants with persistent pulmonary hypertension, a practice not recommended anymore. The obvious clinical benefits of allowing this physiologic extracellular volume contraction to occur, especially in the critically ill newborn, clearly outweigh the clinical importance of a mild contraction alkalosis that develops after recovery. No specific treatment is needed in such cases, because with the stabilization of the extracellular volume and renal function after recovery, acid-base balance rapidly returns to normal. In the last condition, there is a direct stimulation of Na+ reabsorption coupled with H+ loss in the proximal tubule, and an indirect stimulation of H+ loss in the distal nephron by the increased activity of the renin-angiotensin-aldosterone system. Contraction alkalosis responds to administration of saline to replace the intravascular volume in conjunction with additional potassium supplementation to account for renal potassium wasting. In the other disorders, however, the primary problem of reduced glomerular filtration rate or elevated aldosterone must be treated for the alkalosis to resolve. One of the most commonly encountered clinical scenarios of chronic metabolic alkalosis actually occurs in the form of a mixed acid-base disorder in a preterm infant with chronic lung disease on long-term diuretic treatment. By stimulating proximal tubular Na+ reabsorption and thus H+ loss, distal tubular H+ secretion, and renal ammonium production, the diuretic-induced hypokalemia contributes to the severity and maintenance of the metabolic alkalosis. Furthermore, metabolic alkalosis per se worsens hypokalemia, because potassium moves intracellularly to replace hydrogen as the latter shifts into the extracellular space. Although the serum potassium concentration may be decreased, the serum levels in the newborn do not accurately reflect the extent of total-body potassium deficit because potassium is primarily an intracellular ion, with approximately 98% of the total body potassium being in the intracellular compartment. In addition, the condition is often accompanied by marked hypochloremia and hyponatremia. Hyponatremia occurs in part because sodium shifts into the intracellular space to compensate for the depleted intracellular potassium. In this situation, potassium chloride, and not sodium chloride supplementation, reverses hyponatremia and hypochloremia, corrects hypokalemia and metabolic alkalosis, and increases the effectiveness of diuretic therapy. Because chloride deficiency is the predominant cause of the increased pH, ammonium chloride or arginine chloride also corrects the alkalosis. These agents do not affect the other electrolyte imbalances such as the hypokalemia, so they should not be the only therapy given. It is important to keep ahead of the potassium losses in infants receiving long-term diuretic therapy, rather than to attempt to replace potassium after intracellular depletion has occurred. Because the rate of potassium repletion is limited by the rate at which potassium moves intracellularly, correction of total body potassium deficits can require days to weeks. In addition, there is also a risk of acute hyperkalemia if serum potassium levels are driven too high during repletion, particularly in newborns in whom an acute respiratory deterioration may occur, with worsened respiratory acidosis and the subsequent movement of potassium from the intracellular to the extracellular space. The routine use of potassium chloride supplementation and close monitoring of serum sodium, chloride, and potassium levels are therefore recommended during long-term diuretic therapy to prevent these common iatrogenic problems. Respiratory Alkalosis When a primary decrease in Paco2 results in an increase in the arterial pH beyond 7. The initial hypocapnia is acutely titrated by the intracellular buffers, and metabolic compensation by the kidneys returns pH toward normal within 1 to 2 days (see Table 31-5). Interestingly, respiratory alkalosis is the only simple acid-base disorder in which, at least in adults, the pH can completely be normalized by the compensatory mechanisms (Brewer, 1990).
Discount evista 60mg free shipping
However breast cancer nike elite socks cost of evista, a recent study suggests that the etiology may be a maternally inherited, homoplasmic m. The form also associated with cardiomyopathy may be a variant of the benign isolated myopathy and involves striated muscle in both skeletal and cardiac muscle. It manifests in the newborn period with lactic acidosis and a cardiomyopathy that improves during the first year of life. More attention must be paid to these two disease entities, because with early optimal medical care, affected infants may have an excellent prognosis. Lethal Infantile Mitochondrial Disease Infants with lethal infantile mitochondrial disease are severely ill in the first few days or weeks of life or in the extended newborn period. He was delivered by cesarean section because of variable decelerations, and he emerged without meconium staining or passage. An echocardiogram showed concentric right ventricular hypertrophy with elevated right ventricular pressure (70 mm Hg). Initial laboratory studies showed acute metabolic acidosis (blood lactate >18 mmol/L; arterial pH, 6. A left quadriceps muscle biopsy was used for histochemical analysis and to prepare a 10% extract for respiratory chain studies. A series of echocardiograms documented biventricular, hypertrophic, nonobstructive cardiomyopathy. The patient died on the seventh hospital day after a sudden episode of hemoglobin desaturation. The right ventricle was found to be thickened and enlarged, with relative sparing of the left side. The pulmonary vasculature of the lungs showed hypertrophy that extended to the most distal vessels. This baby had a fatal syndrome defined clinically by prenatal cardiomyopathy and severe pulmonary hypertension in the newborn period. This case is an example of a mitochondrial metabolic disorder, but without a specific molecular genetic cause. A more extensive evaluation by current methods might or might not have revealed a diagnosis. Death often occurs by 6 months of age and almost always is associated with overwhelming lactic acidosis. Skeletal muscle shows lipid and glycogen accumulation and abnormally shaped mitochondria on electron microscopic examination. Generalized proximal renal tubular dysfunction may occur, leading to the renal Fanconi syndrome. This disease is characterized as a progressive neurodegenerative disorder with severe hypotonia, seizures, extrapyramidal movement disorders, optic atrophy, and defects in automatic ventilation or respiratory control (Finsterer, 2008; Leigh, 1951). There is no effective treatment for this disease, unless the cause is a specific inability to synthesize coenzyme Q10. It is possible that most of the patients with Leigh disease have disturbances in nuclear-encoded genes. Some patients may manifest hypertrophic cardiomyopathy, liver dysfunction, and microcephaly. The neuropathologic lesions include demyelination, gliosis, necrosis, relative neuronal sparing, and capillary proliferation in specific brain lesions. Commonly, elevation in blood lactate is only slight to moderate, as well as intermittent, in this diverse group of patients. The only example of such a mutation manifesting in early infancy is the Pearson syndrome. This disorder is systemic and primarily affects the hematopoietic system and pancreas function. The characteristics are severe macrocytic anemia with varying degrees of neutropenia and thrombocytopenia. Bone marrow examination shows normal cellularity, but extensive vacuolization of erythroid and myeloid precursors, hemosiderosis, and ringed sideroblasts. However, patients who are able to recover or who benefit from aggressive therapy may demonstrate other signs of this systemic disorder in late infancy or childhood, such as poor growth, pancreas dysfunction, mitochondrial myopathy, lactic acidosis, and progressive neurologic damage. Unidentified Genetic Defects A number of diseases are believed to be caused by mitochondrial respiratory chain problems, but the specific mutations remain unknown. These disorders constitute the group 4 mutations, or the disorders of unknown inheritance (Shoffner, 1995). Infants and children with this progressive disease experience progressive cerebral cortical damage, sometimes also involving the cerebellum, basal ganglia, and brainstem; in some, liver disease may progress to cirrhosis. The neuropathologic lesions consist of spongiform or microcystic cerebral degeneration, gliosis, necrosis, and capillary proliferation Seizures are prominent, including myoclonus. Commonly the condition is untreatable, because it is relentless and unresponsive to alkali therapy. In addition, acidemia per se can easily cause the coma or impaired cardiac contractility that may be encountered. To further complicate the issues, enzymatic and molecular analyses usually are not immediately available. Decisions regarding management must be individualized, because the mitochondrial dysfunction and resultant pathophysiology can vary among infants. Barth Syndrome Barth syndrome is an X-linked disorder associated with cardiomyopathy, skeletal muscle disease, and neutropenia (Yen et al, 2008). Positional cloning identified a gene for this disorder on Xq28 that encodes for a phospholipid remodeling enzyme, cardiolipin acyl transferase. It has been hypothesized that the organic acid 3-methylglutaconate accumulates because of defective mitochondrial transport. It is possible that if severe cholesterol deficiency can be avoided, affected infants may survive and may be relatively free of cardiomyopathy during childhood. Of diagnostic importance, not all patients with 3-methylglutaconic aciduria have Barth syndrome. A few have isolated leucine-dependent 3-methylglutaconyl-CoA hydratase deficiency or Costeff syndrome, but most have ill-defined mitochondropathies. These four categories of metabolic diseases involve molecules important in cell membranes and share overlapping clinical presentations. Clinical presentations are heterogeneous, with a broad range of age at presentation and severity of symptoms. Age of onset varies from prenatal to adulthood, and severity can range from severe disability and early death to nearly normal lifestyle and life span. For each condition, interfamilial variability is greater than intrafamilial variability. The genetic and clinical characteristics of conditions in these categories that can manifest in the neonatal period (except Pompe disease, which is addressed in Chapter 22) are also summarized in Tables 23-1 to 23-3. Important presentations that should lead the neonatologist to consider these disorders in the differential diagnosis are as follows: 1. In utero infection-hepatosplenomegaly and hepatopathy, possibly with extramedullary hematopoiesis 2. Neurologic only-early and often difficult to control seizures, hypertonia or hypotonia, with or without altered head size and with or without eye findings 4. Coarse facial features with bone changes, dysostosis multiplex, or osteoporosis 5. Rarely, known family history or positive prenatal diagnosis Only for the last three presentations are these conditions likely to be considered early in the differential diagnosis. Most babies with these conditions are born to healthy, nonconsanguineous couples with normal family histories, and these disorders are usually considered late, if at all, as in Case Study 1. These lysosomal enzymes are responsible for splitting large molecules into simple, low-molecular-weight compounds, which can be recycled. The materials digested by lysosomes and derived from endocytosis and phagocytosis, are separated from other intracellular materials by the process of autophagy, which is the main mechanism whereby endogenous molecules are delivered to lysosomes. The common element of all compounds digested by lysosomal enzymes is that they contain a carbohydrate portion attached to a protein or lipid. Sphingolipids, globosides, gangliosides, cerebrosides, and lipid sulfates all are glycolipids. The different classes of glycolipids are distinguished from one another primarily by different polar groups at C1.
Purchase 60 mg evista fast delivery
Hypothermia can be accomplished with both whole-body and head cooling breast cancer updates purchase evista 60mg visa, although clinical trials of whole-body cooling more effectively achieved a reduction in adverse outcomes. The therapy is most beneficial when initiated as quickly as possible after an insult, with beneficial effects noted when treatment was initiated within 6 hours of birth. The timing of insult in relation to the time of birth is not always obvious, making it difficult to know the actual timing of initiating therapy after the insult. Mild hypothermia therapy should be considered when an infant has required a significant resuscitation after birth, Apgar scores are low (especially a 5-minute score less than 5), fetal or neonatal acidosis are documented on cord or newborn blood gases, and signs of encephalopathy are apparent. In addition, the history of a significant event likely to cause a hypoxic-ischemic insult should trigger a thorough evaluation of the newborn to determine whether hypothermia therapy is indicated. Hypothermia therapy is not currently available at all institutions; however, all delivery services must be able to recognize the indications for therapy, so that a transfer can be initiated as quickly as possible, if necessary. In infants born without a heart rate or any respiratory effort, if resuscitation is performed to the full extent without any response, discontinuation is recommended after 10 minutes. From a review of 13 years of data from a database including 81,603 deliveries, Haddad et al (2000) found that survival with an Apgar score of 0 at 1 minute occurred in 1. Of 33 infants assigned Apgar scores of 0 at both 1 and 5 minutes, 67% died before hospital discharge (Haddad et al, 2000). A recent review of the available literature for infants with Apgar scores of 0 at 10 minutes found that 94% of infants either died or were severely handicapped, whereas 3% were mild or moderately handicapped (Harrington et al, 2007). The need for neonatal resuscitation, even when no signs of encephalopathy are recognized, increases the risk that children will have lower scores on intelligence quotient tests at school age (Odd et al, 2009). This risk arises most likely because resuscitation is a marker for a prior insult. However, a well-performed resuscitation could be critical for a successful recovery. Neonatal care providers have an obligation to ensure that this process is performed as well as possible and that the techniques of resuscitation are evaluated in an objective manner to promote continued improvement. A randomized, controlled trial of delivery-room respiratory management in very preterm infants, Pediatrics 120:322-329, 2007. For optimal coordinated care, however, adequate medical transport needs to be developed and continually refined to enable delivery of patients to regional centers, and for specialized care to be available for and delivered to patients in distress. For centers that serve as a basic or specialized service, there will be times when subspecialty care is required. For those who deliver subspecialty care, there will also be times for most organizations when transfer to a similar level organization may be required for reasons such as capacity or extraordinary care. For hospitals that do not have birthing centers as part of their facility, the patient population, the acuity of the arriving patients, and potentially the ultimate morbidity and mortality of those patients depend on skilled, efficient, and quality neonatal transport. Although transfers to neonatal centers in the 1960s and early 1970s often occurred in an ad hoc manner, such as in a police car or an ambulance. This level of care, however, is not consistent throughout cities, nationwide, and internationally. Many transport services and centers have grown around individual center needs, without clear attention to coordination and regionalization of services. Competitive systems, often located in similar areas or vying for similar patient populations, have resulted in the duplication of services at the ground and air levels, and at times increased risk and cost to patients and providers as part of the efforts to increase patient volume and revenue. Intrafacility transport may be required for specialty services within a particular institution. Interfacility transport between lower and higher levels of service capability can also occur, as well as between relatively equivalent levels of service because of capacity or other issues. Transported patients may be of high acuity, relatively stable, or in various stages of convalescent care. Each type of transport requires anticipatory planning, appropriate staffing, adequate modalities. As noted in the next section, transfer agreements can help to minimize inefficiencies and enable rapid approval and eventually transport of patients. This chapter will review considerations and requirements for neonatal transport; discuss issues involved in transport team operation, including equipment, personnel, mode of transport, and medical-legal issues; and present general and specific topics, including quality improvement opportunities, that might be encountered in a neonatal transport system (American Academy of Pediatrics et al, 2007; Cornette, 2004; Woodward et al, 2007). These stations were located within certain area hospitals, where additional resources were allocated to provide care for premature infants. One consequence of the formation of these stations was the development of equipment and protocols to transport premature infants from other area hospitals to those specialized areas to receive care. In the 1960s and 1970s, as interest in neonatal care grew, so did the number of hospitals offering services for premature infants. To help optimize the care being delivered, the March of Dimes produced Toward Improving the Outcome of Pregnancy in a 1976 report (Committee on Perinatal Health, 1976). The report stratified maternal and neonatal care into levels based on their complexity, and it proposed the referral of high-risk patients to centers with sufficient personnel and resources to provide care. The goal was to create standard definitions so that comparisons of health outcomes, resource utilization, and costs among regional institutions could be made. High-risk maternity patients would be able to actively participate in selecting a delivery service, and businesses would be able to select appropriate health care resources for their employees. The subsequent March of Dimes publication in 1993 Toward Improving the Outcome of Pregnancy: the 90s and Beyond reiterated the importance of regionalized care and further delineated care levels (Committee on Perinatal Health, 1993). This proliferation has blurred many of the original distinctions between various care systems. Whether driven by third party payers or other factors, with various interpretations and applications of what "regional care" means, the results have been the creation of a variety of care options (Lainwala et al, 2007). Limited space and cooperative longitudinal care planning in some tertiary care units have created the necessity for patient transport back to a unit with less acuity or fewer resources once their critical condition has resolved or stabilized (Attar et al, 2005; Donovan and Schmitt, 1991; Lynch et al, 1988). Regionalization guidelines should support the return to the community for patients who no longer need the highest care level. Although third-party payers often drive decision making, transport relationships develop between various institutions either by formalized transfer and preferred provider agreements or by historical and personal relationships (Attar et al, 2005). Transfer agreements can help to define the roles, understanding, and expectations between institutions and the transport service; they also help frequently to detail reimbursement issues. These agreements set the expectations for participating facilities, with the ultimate goal of the timely movement of patients from one facility to another 2007; Woodward et al, 2007). Nonetheless, there is a trend in the United States and elsewhere toward the centralization of perinatal care (Howell et al, 2002; Wall et al, 2004). Cifuentes et al (2003) supported the idea that whenever possible, women in early preterm labor should be moved to the regional hospital rather than transferring the infant after birth. Investigators trying to describe the optimal neonatal unit caring for very preterm infants in Europe were unable to produce a consensus as to what the ideal neonatal unit size and patient volume should be (Van Reempts et al, 2007). Given the wide variability in levels of neonatal care and the inability to predict premature delivery, neonatal transport will continue to be a dynamic process. Although a telephone call from a referring provider to a receiving provider might be the most efficient way to receive advice or notify a receiving provider of a potential transport, there are more centralized and effective means of communication. First, a centralized, easily recalled, advertised, and monitored (24 hours per day, 7 days per week) communication system should be available. Identifying and publishing a centralized access number for immediate access to the transport system or receiving center personnel are imperative for optimal communication and efficiency (Southard et al, 2005). Anyone who has transported or referred patients to systems without centralized access understands the challenges in working through operators, unit clerks, multiple providers, and often multiple services to enable a singular transport. This process is time consuming and often frustrating for the referring provider, and it is often time that could be better spent in direct assessment and care of the patient, rather than on the telephone with repetitive informational transfer. Ideally a centralized transport communication center would enable a referring provider to make a single call to a single number and immediately receive all the services that they might require. Those services include appreciation and recognition of the need for transfer, identification of appropriate hospital and facility, review of medical issues, and determination of required services. Also included would be simultaneous awareness of need for transport by the personnel responsible for arrangement of particular modes of transport, verification of bed capacity, and any other logistic items that may need to take place prior to the transport. A telephone call made to an individual provider requesting transport requires a sequential process from data gathering through acceptance for admission and eventual transport service or modality identification and dispatch. Centralized access to a communication center allows all those functions to occur simultaneously, enabling more rapid transport response and appropriate involvement of all those required for the management of the particular patient (American Academy of Pediatrics Committee on Fetus and Newborn and Bell, 2007; Woodward et al, 2007). In most cases involving neonatal transport, this provider should be a neonatologist. There may be instances, however, when the referring or receiving physicians may request or desire additional medical expertise. For example, a cyanotic newborn with congenital heart disease may be temporarily improved or stabilized well by the referring neonatologist and have additional stabilization direction provided by the receiving medical command physician; however, invaluable additional management and planning might be added by a partnering cardiac intensive care physician. A communication or transfer center can allow for multiple providers to be linked together during an initial referral call to allow the highest level advice to be presented and discussed among the providers.
Discount 60 mg evista otc
Infrequently they can manifest as an acute intoxicating disorder in the newborn period premier women's health henderson nc order evista online. The laboratory findings consist of an elevation in plasma homocysteine values and a normal or decreased methionine value. Often the homocysteine values are not as elevated as in classic cystathionine -synthetase deficiency. If there is a defect in folate or vitamin B12 metabolism that produces secondary impairment in methionine synthetase activity, there may also be megaloblastic anemia. Some patients with a defect in cobalamin metabolism respond to treatment with high doses of intramuscular hydroxycobalamin. It is also possible to retard homocysteine accumulation and restore methionine levels by administering betaine, which can enhance remethylation of homocysteine to methionine through the alternate betaine methyltransferase pathway. Some investigators have also used pharmacologic doses of cobalamin, folate, and pyridoxine to stimulate flux through either the methionine synthetase or the classic homocysteine pathway. Unfortunately, if infants with this type of disorder are not treated early, there is usually a permanent and devastating effect on cognitive and motor function. Classic homocystinemia caused by cystathionine synthase deficiency can be ascertained by expanded newborn screening in a minority of cases. The elevated methionine that is the critical detected metabolite may require several days to become apparent; that is unfortunate because cystathionine synthetase deficiency is a treatable disorder. Expanded newborn screening has altered the landscape for several B12 processing defects. Ascertainment of elevated methylmalonic acid with the C3 acylcarnitine, the C4 dicarboxylacyl carnitine, or both usually allows for earlier diagnosis and treatment, with apparent mitigation of long-term effects. These disorders are more common than classical homocystinemia, the latter having a frequency in most populations of 1 in 200,000 births or less. Its frequency is between 1 in 10,000 to 1 in 25,000 births, depending on where in the United States it is studied. There are areas of the world such as Ireland, Turkey, and Iran in which it is more frequent, and others such as Finland, Japan, and among Ashkenazi Jews in which it is less prevalent. It is important for physicians caring for newborns to be aware of the pitfalls of screening, which are summarized in Chapter 27. This disease, inherited as an autosomal recessive trait, exemplifies the interaction of a gene and the manipulatable environment. The cutoff value for newborn screening differs by jurisdiction, but both phenylalanine value and the phenylalanine:tyrosine ratio are considered when calling a presumptive positive. By 5 to 7 days of age in the untreated patient, the eponymous phenylketone is found in urine, and another side product of phenylalanine accumulation, phenylacetic acid, may impart the characteristic "mousey" odor to urine and the patient. Greater detail about the disorder can be obtained in Scriver et al (2001), but a synopsis will be given here. In the first 6 months of life, the affected babies may have difficulty with feeding and vomiting. In some instances, persistent vomiting has been associated with the diagnosis of pyloric stenosis, for which corrective surgery has been performed, perhaps inappropriately. It may be related to the effect of high phenylalanine levels on the transport of amino acids across the blood-brain barrier and then into brain neurons or glial elements. The plasma levels of tyrosine may be decreased, especially in patients who are not receiving tyrosine supplementation in the special amino acid powder used as a daily nutrient. Older infants often exhibit long tract findings, such as spastic quadriparesis and spastic quadriplegia. The untreated infant demonstrates microcephaly acquired postnatally and may also have severe behavioral problems in addition to a diagnosis of autism. Typical findings consist of elevated serum plasma phenylalanine levels, normal or subnormal plasma tyrosine levels, and increased urinary excretion of phenylpyruvic acid, phenyllactic acid, and phenylacetic acid (rarely measured routinely, but easily seen if urine organic acids are studied). Deviations from normal plasma levels are believed to be associated with chronic, perhaps acute, effects on brain function and testing performance; therefore the diet should be maintained for life. Positive effects have been frequently claimed, but few carefully controlled studies are available. The positive effects of this supplement have variously been attributed to competition with phenylalanine absorption in the gastrointestinal tract, competition for transport into the brain, and repletion of neurotransmitters. Injections or implantations of a reservoir in which there is an enzyme that breaks down phenylalanine in the body are in the early phases of testing. The phenylalanine hydroxylase gene has been cloned and sequenced, and the mutations responsible for most abnormalities in humans are known. In rare cases, patients with hyperphenylalaninemia have severe disease not because of deficiency of the phenylalanine hydroxylase apoenzyme, but because of deficiency of the active cofactor of this enzyme, tetrahydrobiopterin. Several defects in the metabolism of biopterin can produce this type of hyperphenylalaninemia. Patients with biopterin deficiency can have evidence of severe brain damage despite treatment with a low-phenylalanine diet. Brain damage may be related to deficiencies of other neurotransmitters whose synthesis also depends on adequate levels of tetrahydrobiopterin. These various other defects, such as the dihydropteridine reductase, 6-pyruvoyl tetrahydropterin synthetase, and the guanosine triphosphate cyclohydrolase deficiencies, can be ascertained by urinary measurements of neopterin and biopterin in urine. They are responsive to therapy with neurotransmitter replacement and tetrahydrobiopterin. An organic acid is any organic compound that contains a carboxy functional group but no -amino group as in an amino acid. In this section, we consider the disorders of fatty acid oxidation, ketone body metabolism, and lactic acid metabolism as well as the more classic inherited defects in organic acid metabolism. It is now more common to be confronted with an asymptomatic or early symptomatic patient and a good idea of the diagnosis. As discussed earlier, acidosis and encephalopathy are usually the hallmarks of these syndromes when they manifest in the newborn period. Although more than one enzyme defect may result in methylmalonic acidemia, all are inherited as autosomal recessive traits. In a methyl-transfer reaction, homocysteine is converted to methionine; the reaction is catalyzed by a cobalamin-containing enzyme, 5-methyl-tetrahydrofolate-homocysteine methyltransferase or methionine synthase. Therefore vitamin B12 and folate are important vitamins in homocysteine and methionine metabolism. In classic homocystinuria, the deficient enzyme is the cystathionine -synthetase, which catalyzes the conversion of homocysteine to cystathionase, a precursor of cysteine. This pathway is readily operative in the adult, so that cysteine is a nonessential amino acid. In newborn infants, however, synthesis of cysteine does not occur at the same rate as in adults. Adenosylmethionine-dependent methyl transfer may be extremely important in the central nervous system. Most of the patients who come to clinical attention in the newborn period have defects in the remethylation of homocysteine to methionine; therefore these patients may have a severe deficiency of adenosylmethionine in the brain and spinal cord. This enzyme is present in mitochondria, and it depends on adenosyl-cobalamin for activity. Impaired function can result from either a mutation of the l-methylmalonyl-CoA mutase apoenzyme or deficient availability of the adenosyl form of vitamin B12. The latter may result from impaired cellular metabolism of vitamin B12, including defective activity of the enzyme adenosylcobalamin synthetase. Some patients, but not usually newborn infants with methylmalonic acidemia caused by apoenzyme deficiency, are responsive to pharmacologic therapy with vitamin B12.

Cheap evista 60mg otc
During the second and third trimesters menstrual at age 7 generic evista 60mg online, however, postprandial blood glucose levels were strongly predictive of both birthweight and the overall percentage of macrosomic infants. With postprandial glucose values averaging 120 mg/dL, approximately 20% of infants had macrosomia; a 30% rise in postprandial levels to 160 mg/dL resulted in a predicted percentage of macrosomia of 35%. In contrast, Persson et al (1996) showed that fasting glucose concentrations account for 12% of the variance in birthweight and correlated best with estimates of neonatal fat. Similarly, Uvena-Celebrezze et al (2002) found that the strongest correlation was between fasting glucose and neonatal adiposity rather than postprandial measures. The Pedersen hypothesis (Pedersen, 1977) presumes that abnormal fuel milieu in the maternal bloodstream is reflected contemporaneously in the fetal compartment: Maternal hyperglycemia = Fetal hyperglycemia. During the first and second trimesters, differences in size between fetuses born to diabetic and nondiabetic mothers are usually undetectable with ultrasound measurements. This central depositing of fat is a key characteristic of diabetic macrosomia and underlies the pathology associated with vaginal delivery in these pregnancies. Acker et al (1986) showed that although the incidence of shoulder dystocia is 3% among infants weighing more than 4000 g, the incidence in infants from diabetic pregnancies who weigh more than 4000 g is 16%. Finally, despite our emphasis on birthweight, this alone may not be a sensitive measure of fetal growth. Catalano et al (2003) conducted body composition studies on infants born to mothers with diabetes and found that even when appropriate for gestational age, these infants have increased fat mass and percent body fat compared with a normoglycemic control group. The asymmetry index was 30% higher in diabetic offspring than in the controls by age 8 years. These investigators also showed that offspring with diabetes have permanent derangement of glucose-insulin kinetics, resulting in a higher incidence of impaired glucose tolerance. In addition, Dabelea et al (2000) also reported that the mean ll Maternal fasting blood glucose levels decline from approximately 85 mg/dL to 75 mg/dL. At night, maternal glucose levels drop markedly as the fetus continues to draw glucose stores from the maternal circulation. Postprandial peaks in maternal blood glucose rarely exceed 120 mg/dL at 2 hours or 130 mg/dL at 1 hour. If maternal glucose levels surge excessively after a meal, the consequent fetal hyperglycemia is accompanied by fetal pancreatic beta-cell hyperplasia and hyperinsulinemia. Fetal hyperinsulinemia, lasting only episodically for 1 to 2 hours, has detrimental consequences for fetal growth and well-being, in that it (1) promotes storage of excess nutrients, resulting in macrosomia, and (2) drives catabolism of the oversupply of fuel, using energy and depleting fetal oxygen stores. Providers were blinded to test results if the fasting value was 105 mg/dL or less and the 2-hour plasma glucose level was 200 mg/dL or less. Langer et al (1994) compared the outcomes of diabetic pregnancies managed conventionally (four blood glucose measurements per day) or intensely (seven blood glucose measurements per day). Fasting blood glucose values were maintained between 60 and 90 mg/dL and 2-hour postprandial values at less than 120 mg/dL. The rate of infants born weighing more than 4000 g was 14% in the conventionally managed group, 7% in the intensely managed group, and 8% in nondiabetic controls. Thus, like the reduction of congenital anomalies in mothers with diabetes by means of first-trimester euglycemia, strict glycemic control in the second and third trimesters may reduce the fetal macrosomia rate to near baseline. Fetal Hypoxic Stress As noted previously, episodic maternal hyperglycemia promotes a fetal catabolic state in which oxygen depletion occurs. For example, the drop in fetal oxygen tension causes stimulation of erythropoietin, red cell hyperplasia, and elevation in fetal hematocrit. Profound episodic hyperglycemia in the third trimester causing severe fetal hypoxic stress has been theorized as the cause of sudden intrauterine fetal demise in poorly controlled diabetes. Prevention of Macrosomia Because macrosomic fetuses are at an increased risk for immediate complications related to birth injury and for potential long-term consequences such as late childhood obesity and insulin resistance, measures for prevention of macrosomia have been recommended. As described previously, fetal hyperinsulinemia, which acts as a fetal growth factor, occurs in response to fetal hyperglycemia, which in turn reflects the maternal hyperglycemic condition. Therefore, measures that promote consistent maternal euglycemia may prevent macrosomia. This nomenclature is useful because it categorizes patients according to the underlying pathogenesis of their diabetes-insulin-deficient (type 1) and insulinresistant (type 2 and gestational). One must remember that the diagnosis of gestational diabetes applies to any woman who is found to have hyperglycemia during pregnancy. A certain percentage of such women actually have type 2 diabetes, but the diagnosis cannot be confirmed until postpartum testing. Pregestational Diabetes Patients with type 1 diabetes mellitus typically exhibit hyperglycemia, ketosis, and dehydration in childhood or adolescence. Often the diagnosis is made during a hospital admission for diabetic ketoacidosis and coma. Conversely, it is not unusual for women with a tentative diagnosis of gestational diabetes to be found to have overt, type 2 diabetes mellitus after delivery. The American Diabetic Association has outlined three criteria for diagnosing type 2 diabetes mellitus in nonpregnant subjects. However, the Fourth International Workshop-Conference on Gestational Diabetes and the American College of Obstetricians and Gynecologists have now indicated that either a risk-factor approach or universal screening can be considered (American College of Obstetricians and Gynecologists, 2001; Metzger and Coustan, 1998). Therefore in patients meeting all criteria listed in Table 9-3 and Box 9-2, it may be cost effective to avoid screening. Casual is defined as any time of day without regard to time of last meal, or Fasting glucose level >126 mg/dL (7. Fasting is defined as no caloric intake for at least 8 hours, or Two-hour plasma glucose >200 mg/dL (11. Adapted from the American Diabetes Assoication: Clinical practice recommendations: Standards of medical care for diabetes, 2007, Diabetes Care 30:s4-s41, 2007. Maternal age >25 years Previous infant >4 kg Previous unexplained fetal demise Previous pregnancy with gestational diabetes Strong immediate family history of type 2 or gestational diabetes mellitus Maternal obesity (>90 kg) Fasting glucose level >140 mg/dL (7. Universal screening should be performed in women in ethnic groups at a higher risk for glucose intolerance during pregnancy, namely those of Hispanic, African, Native American, South or East Asian, Pacific Islands, or Indigenous Australian ancestry. For simplicity of administration, universal screening, with the possible exception of the lowest risk category, is probably best. Because maternal insulin resistance rises progressively during pregnancy, screening too early can miss some patients who will become glucose intolerant later. Screening too late in the third trimester can limit the time during which metabolic interventions can take place. Factors that should lead to a first-trimester glucose challenge test are listed in Box 9-3. Various threshold levels for the 50-g glucose challenge are in use, including 140 mg/dL, 135 mg/dL, and 130 mg/dL. This is also why every patient who delivered an infant with macrosomia in a prior pregnancy should be screened early in all subsequent pregnancies. Either 100 g of glucose and 3 hours of testing, or 75 g of glucose and 2 hours of testing can be used. Nevertheless, the currently reported perinatal mortality rates among women with diabetes remain approximately twice those observed in nondiabetic women (Table 9-4). In the past decade, fewer intrauterine deaths have been reported, probably reflecting more careful fetal monitoring. Shoulder dystocia, defined as difficulty in delivering the fetal body after expulsion of the fetal head, is an obstetric emergency that places the fetus and mother at great risk. Widness et al (1990) demonstrated that hyperglycemia is a powerful stimulus to fetal erythropoietin production, probably mediated by decreased fetal oxygen tension. Neonatal polycythemia may promote vascular sludging, ischemia, and infarction of vital tissues, including the kidneys and central nervous system. This complication is usually much milder and less common in the infant of a woman whose insulindependent diabetes is well controlled throughout the entire pregnancy and who exhibits euglycemia during labor and delivery. Mortality Fetal Neonatal Perinatal *California data for 1986, corrected for birthweight, sex, and race. Although half of shoulder dystocias occur in infants of normal birthweight (2500 to 4000 g), the incidence of shoulder dystocia is 10-fold higher (5% to 7%) among infants weighing 4000 g or more and rises to 31% for infants whose mothers have diabetes (Gilbert et al, 1999; see also Chapter 15 for a discussion of complicated deliveries and Chapter 64 for a discussion of the neurologic consequences of birth injury. Increased destruction of red blood cells contributes to the risks of jaundice and kernicterus. This complication is usually managed using phototherapy, but exchange transfusions may be necessary for marked bilirubin elevations. Hypertrophic and Congestive Cardiomyopathy In some infants with macrosomia of mothers with poorly controlled diabetes, a thickened myocardium and significant septal hypertrophy have been described (Gutgesell et al, 1976). Although the prevalence of myocardial Neonatal Morbidity and Mortality For a complete discussion of neonatal morbidity and mortality, see Chapter 94. Echocardiograms show a hypercontractile, thickened myocardium, often with septal hypertrophy disproportionate to the ventricular free walls.
