

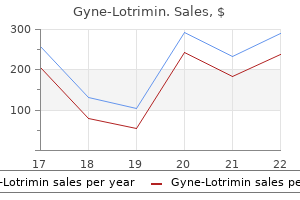
Buy cheap gyne-lotrimin 100 mg on line
Housing and workplace modifications can allow patients to maintain independence despite worsening disability womens health sex purchase 100 mg gyne-lotrimin overnight delivery. Therapeutic discoveries from animal models and clinical research raise the prospect for new treatments in the foreseeable future. The function of this gene is currently unknown, though the encoded protein has been shown to be involved in the initiation of autophagy. These variants were not found in normal controls, but linkage with the disease was not established. Motor neurons are large cells with high metabolic requirements, and mitochondrial dysfunction and oxidative stress are likely to be involved in their susceptibility to degeneration. They have numerous glutamatergic inputs, and are therefore vulnerable to excitotoxicity. Motor neurons have the longest axons of any human cell and some evidence suggests that the disease process begins at the distal axon, perhaps due to perturbed axonal transport and disruption of the cytoskeleton. Intracellular protein aggregation is a notable pathological feature of the disease, although it is not known whether this is damaging to the cell. There has also been recent interest in the role of the interactions between motor neurons and glial cells and inflammatory events in the pathogenesis of the disease. There is increasing evidence from case-control studies to support physical activity as a risk factor in susceptible individuals, but further population-based studies are needed, and it remains unclear whether any associations are causative or co-occur with an as-yet uncharacterized risk profile. Pathology Lower motor neurons degenerate in clinically affected areas of the spinal cord and brain stem, and associated diffuse astrocytic gliosis occurs. Other inclusion bodies, such as eosinophilic Bunina bodies and accumulations of neurofilaments, are also seen. Axonal loss and gliosis in the descending motor pathways gives rise to the lateral sclerosis, most prominent in the medulla and cervical spinal cord, which gives the disease its name. For example, changes in the cerebellum, basal ganglia and sensory areas have been identified in imaging studies and abnormalities of central and peripheral sensory pathways described in electrophysiological studies. More subtle cognitive impairment is evident on detailed neuropsychological testing in a much higher proportion of patients. Patients tend to present with symptoms in a single body region, bulbar, arm or leg in approximate thirds and respiratory in approximately 1%. With time, the disease usually generalizes and symptoms in multiple regions become apparent. In routine clinical practice, most diagnoses are made at these earlier, incomplete stages. Asymptomatic involvement of other limbs, especially fasciculations, is often evident on examination. Wasted, fasciculating muscles with brisk or even retained reflexes are an ominous finding, especially if present in the bulbar region or more than one body region. With progressive disease, patients become wheelchair or bed-bound, or unable to use their arms for activities of daily living. Some people choose tube feeding to prevent an intolerable situation of hunger that cannot be alleviated due to inadequate bulbar function. The cough becomes weak because of vocal cord paresis and weakness of expiratory muscles and this further increases aspiration risk. Weight loss is a common feature, can partly be attributed to bulbar dysfunction and muscle loss but also appears to reflect a direct hypermetabolic effect of the disease process. Weakness of the diaphragm and intercostal muscles almost invariably develops as the disease progresses. With the onset of respiratory failure, patients may complain of dyspnoea or orthopnoea, but more subtle symptoms are often earlier manifestations, such as fragmented sleep, daytime somnolence, anorexia, and morning headaches due to nocturnal carbon dioxide retention. These symptoms may not be volunteered by patients and should be specifically sought, as they are treatable with noninvasive ventilation. Death commonly results from ventilatory respiratory failure or aspiration pneumonia. Median survival from first symptom onset in bulbar-onset disease is 20 months and, in limb-onset disease, 29 months. Fifteen per cent (15%) of patients with limb-onset disease survive longer than 5 years. Fasciculations can be normal in athletes and benign crampfasciculation syndromes without evidence of weakness or denervation, usually affecting middle-aged adults, do not evolve into motor neuron disease. Multifocal motor neuropathy is an important differential of progressive muscular atrophy because it is treatable. There may be marked weakness but little wasting, predominantly affecting the arms. Proximal conduction block can be difficult to identify on electrophysiology and lumbar puncture and a trial of intravenous immunoglobulin may be warranted in suspected cases. Genetic analysis of the androgen receptor gene should be performed in suspected cases. More commonly, there are additional features such as cerebellar ataxia or dementia. Hexosaminidase assays should be reserved for young and/or atypical patients, particularly in those of Ashkenazi Jewish extraction. Giving the diagnosis Time is required because the news is devastating and patients and their families often have many difficult questions. A skilled clinician will convey the necessary hard truths about implications and prognosis while providing support and maintaining hope. Ongoing and active support following diagnosis is very important, and can be most effectively provided in specialist centres by a multidisciplinary team. Electrophysiology is performed to confirm active and chronic denervation in multiple regions and to exclude potentially treatable mimics, such as demyelinating neuropathy, myopathy, or myaesthenia gravis (the latter especially in patients with bulbar onset). Riluzole, an antiglutamatergic agent, remains the only conclusively proven disease-modifying therapy. There were small beneficial effects on both bulbar and limb function, but not on muscle strength. Riluzole is generally well tolerated by patients; nausea, gastrointestinal upset, and raised transaminase enzyme levels may occur but are often transient and self-limiting. Clinical trials of many different agents including branched-chain amino acids, dextromethorphan, total lymphoid irradiation, the free radical scavenger acetylcysteine, gabapentin, creatine, vitamin E, lithium, coenzyme Q10, olesoxime, pentoxyfilline, glatiramer acetate, dexpramipexole have all proven negative to date. There was no effect on survival and the medication is not currently licensed for use in Europe. Symptomatic therapy In contrast to the limited disease-modifying therapeutic options, much can be done to address symptoms, disability, and distress. A multidisciplinary setting appears to improve survival in observational studies and usually includes a neurologist, specialist nurse, respiratory, anaesthetic and gastroenterology expertise (for noninvasive ventilation and gastrostomy insertion), speech and language therapist, physiotherapist, occupational therapist, social worker, and links to palliative care medicine. Charities, such as the Motor Neuron Disease Association in the United Kingdom, are often able to facilitate equipment provision. Respiratory function can be monitored clinically, and through forced/slow vital capacity, noninvasive random capnography and capillary blood gases, and augmented by overnight oximetry and capnometry when necessary. Cough assist devices are used to help clear respiratory secretions, and have hypothetical advantages in preventing aspiration pneumonia; a clinical trial is required to assess whether there is any survival benefit. Many patients prefer, and many clinicians advise, to avoid endotracheal intubation in a disease causing such widespread and irreversible weakness, as a locked-in syndrome is the end result. Excess salivation can be a significant problem, especially for patients with bulbar disease, and can be effectively treated using anticholinergics, such as hyoscine patches, sublingual atropine drops, glycopyrronium, amitriptyline tablets, or botulinum toxin injection to the salivary glands. Early swallowing problems may be addressed by simple measures such as a chin tuck, attention to food consistency, and nutritional supplements. Later, more severe dysphagia can be treated with a gastrostomy tube inserted either under endoscopic or radiological guidance. Some centres now perform a hybrid technique, which allows a larger diameter, more secure tube to be placed without conscious sedation, and in the presence of noninvasive ventilation, an important consideration as gastrostomy insertion becomes more risky with respiratory weakness, especially when forced vital capacity falls below 50%. Patients and clinicians may consider gastrostomy in the presence of significant weight loss, recurrent aspiration pneumonia, or when frequent choking or dysphagia makes mealtimes prolonged or intolerable. The aims are to avoid a hungry, dysphagic patient unable to fulfil their nutritional requirements and to facilitate discharge home. Multifocal motor neuropathy with conduction block Patients may present at any stage of adult life with multifocal and slowly progressive muscle weakness over as much as 20 years.
Generic gyne-lotrimin 100mg line
They comprise several disorders with different molecular pathological bases for the diseases breast cancer diet discount 100 mg gyne-lotrimin with visa. In all of these conditions, serum creatine kinase is either normal or much lower than seen in many congenital muscular dystrophies. With later childhood presentation the differential diagnosis is as already mentioned, plus Duchenne muscular dystrophy (though calf hypertrophy is usually more pronounced and serum creatine kinase is typically higher-biopsy will exclude the diagnosis) or childhood presentation of a limb-girdle type of muscular dystrophy. Classification There are several recognized forms of congenital muscular dystrophy and, as there is considerable heterogeneity in the group that remains, additional entities are ultimately likely to be distinguished at the genetic level. The diagnostic classification of this group of diseases was previously very clinically based, but is moving increasingly into a molecularly based. If they survive this period, with appropriate management of feeding problems, and respiratory and (in a minority) cardiac complications, survival into adult life is the norm. The muscle weakness in congenital muscular dystrophy may be relatively static, but the complications of that weakness can be severe, and vary according to the precise diagnosis. This investigation can be carried out on a variety of tissues including skin, muscle, and placenta. Mutation testing Characteristic pattern of joint hyperlaxity distally with proximal contractures. Feeding problems may be intractable and lead to chronic malnutrition unless treated by nasogastric or gastrostomy feeding. Malnutrition may contribute to susceptibility to chest infections, which is also heightened by weakness of the respiratory muscles. These children are at risk of respiratory failure and their follow-up should include monitoring for this complication, which can be effectively managed by the provision of noninvasive home nocturnal ventilation. Manifesting carriers of Duchenne muscular dystrophy/Becker muscular dystrophy A highly variable group, which may occasionally be as severely affected as those with Duchenne muscular dystrophy or more or less mildly than those with Becker muscular dystrophy. It is, therefore, important to recognize dominant acting mutations-segregation studies are needed to determine whether the mutation is de novo, but the possibility of somatic or germline mosaicism in de novo mutations should always be considered to provide correct counselling. As the molecular basis for these disorders has recently become much better established, specific diagnosis should be attempted in all cases in order to allow proper direction of management, as well as prenatal and carrier testing where requested. Formal examination of a small child may be difficult, and the main clinical tool is observation of walking, attempting to run, jump, and climb stairs, and to rise from the floor. It is imperative to give the child space to attempt to run, as this will bring out the lack of spring in the step and the lack of fluidity of the attempted running. They frequently Dystrophin deficiency this group, including two of the most common forms of muscular dystrophy-Duchenne and Becker muscular dystrophy-involve the same gene and protein. For patients in whom deletion and duplication analysis are negative, testing for point mutations via direct sequencing techniques (Sanger sequencing or next-generation sequencing) of the entire coding region is mandatory. This analysis also allows the distinction of dystrophinopathy from the much rarer (in most populations) limb-girdle types of muscular dystrophy. Precision of the exact mutation is important for offering carrier testing to the mother and other family members (important to establish the risk of any cardiac problems as well as to allow genetic advice for future pregnancies) and also to allow future access to the mutation-specific treatments that are currently under development, such as antisense oligonucleotide-mediated exon skipping and stop codon suppression. These patients are susceptible to cardiac failure at any age from the teens onward and should be monitored for this complication on a regular basis (Box 24. It is frequently asymptomatic, and needs to be sought through full cardiac assessment including echocardiography, as treatment with antifailure medication may improve function and prognosis. This young man has now been maintained on home nocturnal ventilation successfully for more than 7 years. Note hypertrophic muscles in calves and quadriceps and mild wasting around the shoulder girdle. In normal muscle, dystrophin labels evenly around the periphery of the muscle fibres. This labelling is typically patchy and reduced in Becker muscular dystrophy, and is either completely or almost completely absent in Duchenne muscular dystrophy. Serum creatine kinase is always massively elevated, even to more than 200 times normal, but levels of serum creatine kinase do not distinguish the severity of the disease. Muscle biopsy and electromyography are nonspecifically but generally severely dystrophic. Molecular confirmation of the diagnosis is essential to assist in defining prognosis and to provide appropriate genetic counselling. In addition, current standards of diagnosis include detailed characterization of the mutation as it is essential in view of mutation-specific drugs that are now available or being developed for use, so far, in clinical trials. Over the whole group, there is a correlation between dystrophin abundance (as measured in a muscle biopsy sample) and severity: children with completely absent dystrophin tend to be confined to a wheelchair slightly earlier than children whose biopsies contain low levels of dystrophin. Although these correlations are useful in a general sense, they are not absolutely predictive of outcome in an individual case, and must always be taken in the context of the clinical features of the patient. Indeed, the move towards genetic testing as the primary step in diagnostics means that most patients now will not have a muscle biopsy routinely. They can be useful, however, in giving the best possible guide to prognosis, especially in those patients who present early with no clinical clues as to the severity of the disease, or who are identified by neonatal screening or the incidental finding of a high serum creatine kinase level. Once the diagnosis has been considered, measurement of the serum creatine kinase will confirm the suspicion and ideally a referral to a specialist unit should be made at this stage. As part of the multidisciplinary management, the introduction of nocturnal noninvasive ventilation and scoliosis surgery has been shown to have a positive impact on survival, in particular if combined. The key to proper management is a team specializing in neuromuscular management, who can oversee the coordination of input from physiotherapy, orthopaedic, cardiac, respiratory and psychology specialists. Input from other specialties, including occupational therapy, educational psychology, gastrointestinal medicine, and palliative care, may also be required. The current mean age at diagnosis has improved over the last decade dropping from almost 5 years to 4. The principal impetus to early diagnosis is currently the ability to offer parents the option of prenatal diagnosis in subsequent pregnancies. Early diagnosis is essential, as prompt management and interventions according to standards of care results in better outcomes. All of their daughters are obligate carriers of Becker muscular dystrophy, but none of their sons are at risk. Corticosteroids are at present the sole available treatment that has been proven to slow down the decline in muscle strength and prolong ambulation. It is recommended that corticosteroids (prednisolone or deflazacort) are started between the age of 2 and 5 years (when strength is plateauing or declining) for the preservation of strength. An increasing consensus has developed that use of daily corticosteroids (prednisolone at a dose of 0. Controlled trials show a clear benefit for strength and respiratory function for up to 18 months, and uncontrolled long-term cohort studies report a significant delay in loss of ambulation, a reduction in the development of scoliosis, and a protective effect on respiratory and also cardiac function. Close monitoring for the side effects of corticosteroids is, of course, indicated, and crucial to minimize the potentially harmful effects of the treatment. The prophylactic use of bisphosphonates in this patient group remains controversial, but intravenous bisphosphonate treatment is certainly indicated if there is a problem with symptomatic fracture, and this need not be an indication for discontinuation of steroid treatment. Duchenne muscular dystrophy: after mobility is lost Despite optimal multidisciplinary management loss of ambulation inevitably occurs and interventions and adaptation should follow according to disease progression (early and late nonambulatory phase). The prompt provision of a powered wheelchair with indoor and outdoor access is critical to the best possible maintenance of independence and access to wheelchair sports. In the early nonambulatory phase provision of a manual wheelchair may also be necessary, predominantly for indoor use and as a backup. With disease progression upper limb strength will inevitably decline and alternative control devices need to be considered to maintain independence. Regular assessment of the wheelchair, with particular attention to correct seating, promoting upright and symmetrical spine posture, is fundamental to reduce scoliosis, as well as lower limb neutral posture to limit foot deformities. For patients with progressive scoliosis, spinal surgery in an experienced setting is a good way to restore posture and comfort, and has an additive effect on survival. Physiotherapy priorities for all boys shift towards postural support, the prevention and containment of contractures, and respiratory maintenance.
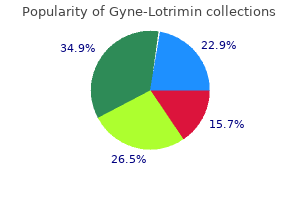
Buy gyne-lotrimin 100 mg online
For mild to moderate anxiety women's health clinic san antonio generic gyne-lotrimin 100 mg overnight delivery, either modality may be effective as a first-line treatment. For patients with moderate to severe anxiety, pharmacotherapy on a short- or longterm basis is usually needed. Serotonin reuptake inhibitors the serotonin reuptake inhibitors are now first-line treatment for anxiety disorder. There is no clear difference in efficacy among these agents, but side effect profiles differ. Many anxious patients are highly sensitive to medications, so it is important to start slowly and provide education about the frequency of side effects and time course of response, in order to avoid premature discontinuation. Many patients will experience initial gastrointestinal discomfort including nausea, but it is important to reassure the patient that this side effect disappears for almost everyone within the first week of treatment. They should be told to take the medication with food, at least until the gastrointestinal side effects have remitted. For some patients, it may be beneficial to start at a lower than usual starting dosage and titrate up once the gastrointestinal side effects abate. Sexual side effects include decreased interest and anorgasmia for women and erectile or ejaculatory dysfunction for men. These side effects do not dissipate but are fully reversible upon discontinuation. If after four to six weeks of treatment there has not been substantial improvement, the dosage can be increased. If there is still no improvement, alternative agents or psychotherapy may be needed. Patients need to be educated not Withdrawal may cause anxiety missed medical diagnoses in patients with concurrent anxiety by highlighting those symptoms. It is important to note that anxiety commonly coexists with depression and that all anxiety patients should be assessed for depression. Diagnosis When a patient is anxious, it is important to first ask how persistent and severe the anxiety is. For patients whose level of functioning is poor, also asking about functional level prior to the onset of symptoms can help establish the extent to which anxiety is contributing to functional impairment. Treatment When a medical patient is anxious, the first step is to ask them why and see if information and addressing their fears, for example, about prognosis, is effective. Feeling nervous, anxious, or on edge Not at all = 0 Several days = 1 More than one-half of the days = 2 Nearly every day = 3 Not at all = 0 Several days = 1 More than one-half of the days = 2 Nearly every day = 3 2. Being unable to stop or control worrying Two-item total score of greater or equal to 3 represents a positive screen. Pharmacologic treatment for panic disorder should continue for at least six months after the symptoms have resolved. For patients with chronic or recurrent panic disorder, or years of generalized anxiety disorder, treatment may continue indefinitely. Some patients may be reluctant to taper off if there are ongoing severe psychosocial stressors. Other agents Benzodiazepines, while still widely used, should not usually be first line for an anxiety disorder as they can cause sedation and cognitive dysfunction, are addictive, and commonly abused. However, they are useful for short-term anxiety, such as that associated with a medical procedure. The serotonin and norepinephrine reuptake inhibitors venlafaxine and duloxetine are usually effective alternatives for anxiety. They may be preferentially indicated for patients with comorbid pain such as neuropathic pain or fibromyalgia. Psychological treatments Where available, psychotherapy is the treatment of choice for many patients with anxiety disorders. The physician can offer brief psychological interventions themselves that are often effective, particularly for those with mild to moderate anxiety. For example, they may encourage patients to question fearful thoughts, replacing undue fears with more realistic appraisals, and listing the worst possible scenarios in order to help the patient see that they can handle feared future events. Referral for specialized treatments Patients should be referred for consultation or treatment with a psychologist or psychiatrist for severe or refractory symptoms, patient preference, or selected disorders that are generally beyond the expertise of the primary care physician (Table 26. Specific phobias such as snake or bridge phobias rarely present in medical settings, but needle or pill-swallowing phobias may interfere with medical care. These trauma-related disorders are not only an important cause of suffering but may also complicate medical care, hence they require recognition and appropriate treatment. Introduction There is a group of psychiatric disorders specifically defined as being a response to severe psychological stress or trauma. Acute stress disorder is the immediate emotional and behaviour reaction to an acute stress, for example, being told that a family member has died unexpectedly. Adjustment disorders are longer-lasting emotional and behavioural reactions to ongoing stressors, such as a new diagnosis of cancer. Accidents and acute illness may be traumatic, as on occasion may treatment itself. These disorders are not only an important cause of suffering but may also significantly complicate medical care, for example, by leading to the avoidance of essential medical treatment. Overall, approximately half of patients with anxiety disorders respond fully to either drug or psychological treatment and the remainder respond to varying degrees, but the relapse rate is high. Consequently many hospitalized patients may be considered to have suffered trauma, but most people exposed to traumatic events do not go on to develop a mental disorder; they are surprisingly resilient in the face of adversity. However, many people who have suffered traumatic events become severely distressed and some go on to develop a mental disorder. Because trauma is so common in medical practice, in the form of accidents, severe illness, and sometimes medical and surgical treatments, these disorders are commonly seen by physicians. An initial severe reaction to a traumatic event such as severe accident is an acute stress disorder and is commonly characterized by dissociation. A more long-lasting emotional reaction to ongoing stress such as a new diagnosis of life-threatening illness is termed an adjustment disorder. An often longer-lasting and more severe psychological reaction Epidemiology the point prevalence of acute stress disorder following exposure to traumatic events is between 5 and 20%, depending on the type Box 26. The point prevalence of adjustment disorders in the general population is between 3 and 12%, with a much higher prevalence in medical populations. Avoidance Acute stress disorder this is an immediate response to a severe stressor. A person with acute stress disorder typically reports experiencing the world as unreal or dreamlike, feeling detached from their body, or that they are having increasing difficulty recalling specific details of the traumatic event (dissociative amnesia). The traumatic event may be persistently re-experienced as recurrent images, thoughts, or dreams. There may also be avoidance of stimuli that arouse recollections of the trauma. They may also suffer from anxiety or symptoms associated with increased arousal such as difficulty sleeping, irritability, and poor concentration. The symptoms of acute stress disorder must last for a minimum of three days and a maximum of four weeks, and must occur within four weeks of the traumatic event. The person may cope with their emotional reaction by misuse of drugs or alcohol, which complicates the clinical picture. Negative alterations in cognitions and mood Marked alterations in arousal and reactivity Adjustment disorders these disorders are a longer-term response to ongoing stressors of a range of severities. The symptoms of adjustment disorders are varied and often include low mood, anger, anxiety, and sleep disturbance. These symptoms must be out of proportion to the severity or intensity of the stressor and be associated with significant impairment in social, occupational, or other important areas of functioning. They occur within three months of the onset of the stressor(s) and do not last for longer than six months after the stressor and its consequences have ended. Assessment and differential diagnosis In assessing individuals, the main differential diagnosis to consider is a normal reaction to a traumatic event. Distress can be a normal, healthy reaction following a stressful event and it is factors such as the intensity and duration of symptoms along with impact on functioning that are important to consider in determining whether a reaction is pathological or not. These conditions must also be differentiated from other psychiatric diagnoses including depressive disorders and anxiety disorders. Adjustment disorder is defined as symptoms that do not last longer than six months after the stressor and its consequences have ended.
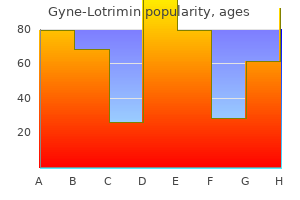
Purchase gyne-lotrimin on line
Referring to smaller-scale variants breast cancer bake sale ideas purchase 100mg gyne-lotrimin amex, these can be described as either in-frame or out-of-frame depending on whether the number of deleted/inserted nucleotides is a multiple of three. If it is, the overall reading frame of the gene is maintained (as the code is read in triplets) and a protein is synthesized even though there will be loss or introduction of amino acids in the polypeptide chain. If it is not a multiple of three, the reading frame shifts and the process is deranged. However, it is not clear whether this is a result of the technique itself or the underlying aetiology of infertility (28, 29). Applied cytogenetics the technique of karyotyping, which allows the visualization of chromosomes of cultured cells that are arrested in metaphase, has been the mainstay of cytogenetic investigations. Chromosomes are stained to produce a characteristic banding pattern with Giemsa stain being widely used (G-banding) (30, 31). Its usefulness is limited by its sensitivity which depends on the size of the chromosomal segment involved and the skill of the operator. Though the technique has evolved and its resolution has improved over the years, it can still only detect abnormalities which are about 5 million base pairs (Mb) in length or greater. The technique is particularly useful in identifying balanced chromosomal rearrangements such as translocations or inversions even if these rearrangements do not result in the net loss or gain of genetic material, an advantage over newer molecular techniques that rely on quantification rather than visualization. In the context of obstetric practice, the karyotype remains the basis of genetic assessment of a fetus following interventional prenatal techniques such as chorionic villous sampling and amniocentesis. It is also the technique of choice in investigating couples presenting with recurrent early pregnancy losses or who have had a baby diagnosed with a chromosomal abnormality based on its ability to detect balanced rearrangements that can themselves predispose to unbalanced chromosome complements in the offspring. Smaller, submicroscopic deletions (microdeletions) cannot be identified using the routine karyotype. In contrast to the karyotype, the clinician needs to ask the laboratory to carry out a targeted test using a specific probe, the implication being that this requires prior clinical suspicion. With the exception of chromosomal aneuploidies, this syndrome is one of the commonest causes of heart defects detected antenatally (34). Epigenetic mechanisms underlie the differential expression of genes in different tissues and, in doing so, underpin development and tissue specification (15). The pattern of selective silencing of certain genes and transcriptional activation of others is mediated by specific protein complexes and is stably inherited through mitotic divisions unless specifically erased and reprogrammed. Gametogenesis and early embryo development are particularly important periods in epigenetic programming. There is strong evidence that imprinted genes play an important role in the regulation of fetal growth, shedding new light on this key aspect of pregnancy (23, 24). The karyotype in (a) has 45 chromosomes as a result of fusion of the long arms of the acrocentric chromosomes 14 and 21 (arrow shows the derivative chromosome). In (b), there is an exchange between the short arms of one chromosome 3 and one chromosome 7. The arrows denote the derivative chromosomes and the break points at bands 3p13 and 7p13. Commonly used platforms identify imbalances which are about 200 thousand base pairs (kb) or longer in length though higher resolutions can be achieved. Assessing the potential pathogenicity of such findings follows a number of steps including parental testing to determine whether this is a de novo or inherited variant. Applied molecular genetics the basis of molecular genetic techniques is their ability to identify the sequence of nucleotides at a specific gene locus and, by comparing this with the normal reference sequence, identify genetic variants which may or may not be relevant to the clinical context in which tests are initiated. The chromosome with the deletion (arrow) exhibits the green signal for the chromosome 22 marker but lacks the red signal for the 22q11. The signals appear paired because chromosomes are in the form of duplicated chromatids at this stage of mitosis. Gene sequencing is the centrepiece of modern molecular genetics with Sanger sequencing being considered as the gold standard (41). Sequencing techniques are not always best placed to detect specific types of mutations and partial or whole gene deletions/duplications require a different approach. Sequencing techniques are also widely used to determine the presence of a specific mutation though this can also be achieved by techniques targeting the specific position within the gene. The sequence data is then contrasted against a library of reference sequences and discrepancies are noted. The efficiency of this technology has made the assessment of the entire exome and genome available for the investigation of disease and has led to the identification of the genetic basis of many monogenic disorders that had eluded molecular characterization (49, 50). Genetic aspects of the clinical encounter the assessment of the inheritance pattern of disorders and traits is by no means restricted to the work of clinical geneticists and a family history is an essential component of most clinical encounters. The relevant information is depicted in the genogram which can be updated as and when necessary and which underpins genetic interpretation. A number of computer programs allow genograms to be generated and updated electronically. It conveys details on diagnoses, causes of death, and adverse pregnancy outcomes and is an accurate record of genetic relationships. The genogram helps establish modes of inheritance for genetic conditions, demonstrates risk patterns, and identifies clusters of events (such as recurrent early pregnancy loss) that are relevant to the consultation. In addition to other cases of ovarian malignancy, particular attention is paid to cancers that point towards the possibility of an underlying cancer predisposition syndrome (breast, endometrial, colorectal). The peaks represent the four bases, each labelled with a different fluorescent dye, with the control sequence represented in (a). Guanine is substituted by adenine in a heterozygous state in (b), where both peaks are seen, and in a homozygous state in (c). The software sequencing numbers shown in this chromatogram do not correspond to base positions and differ between alignments. Issues that feature in the preconception interview include infertility or subfertility, health-promotion advice, the impact of pregnancy on a pre-existing medical condition, and possible genetic risks to the baby. Discussion of most of these issues is beyond the scope of this chapter and the following subsections will concentrate on the approach to possible genetic implications in a future pregnancy. The prospective parents may themselves be affected or concerns may be raised because of a previously affected child, a possible or confirmed genetic diagnosis in the family, a genetic condition that is prevalent in the specific population, the possible implications of consanguinity, and repeated losses of pregnancy or birth of babies with congenital malformations in the family. Whatever the specific concerns, a detailed family history should be taken and a genogram drawn capturing as many relevant details as possible. This will form the basis for subsequent discussion, planning, investigation, and relevant counselling. Affected prospective parents A distinction should be made between monogenic conditions, conditions with multifactorial aetiology and recognized genetic predisposition, and sporadic disease. The rest of the discussion will concentrate on the scenario of prospective parents with single gene disorders. Logical follow-up questions include whether inheritance of the mutation means that the child will develop the condition and if the child will be affected in the same way as the parent. These are key questions in genetics and relate to the concepts of penetrance and expressivity. Penetrance is the percentage of individuals carrying a pathogenic dominant mutation known to cause a condition who will go on to develop the condition regardless of its severity. Expressivity describes the variation in severity of a genetic condition between affected individuals who carry the same mutation (parent and child, for example). On the other hand, this is a condition with very variable expressivity and a mildly affected parent with mostly cutaneous manifestations may have an affected child with complex plexiform neurofibromas or an optic glioma. Another phenomenon relevant to some autosomal dominant conditions is that of anticipation. It describes an increase in severity from generation to generation and is characteristically observed in disorders caused by a type of insertion mutation known as triplet repeat expansion. Instability of the number of triplet repeats during gametogenesis underlies this phenomenon which in some conditions is largely dependent on the sex of the transmitting parent. For example, in Huntington disease, large expansions typically occur in the paternal line (51).
Diseases
- McArdle disease
- Parry-Romberg syndrome
- Pendred syndrome
- Metaphyseal chondrodysplasia Schmid type
- Cold agglutinin disease
- Glycogenosis, type 0
- Bardet Biedl syndrome
- Thrombocytopenia purpura
- Sweet syndrome
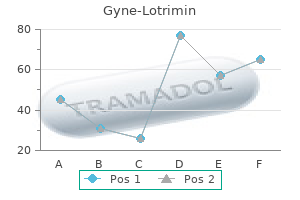
Generic gyne-lotrimin 100mg on line
Injections of botulinum toxin are usually helpful menstruation synonym order gyne-lotrimin canada, but have to be repeated every 3 months. Neurosurgical intervention to relieve compression of the facial nerve by aberrant vessels in the posterior fossa may help selected cases. The condition must be distinguished from the very common benign fasciculations of the face, usually periorbital, related to fatigue or emotional tension, and from facial myokymia that is occasionally encountered as a manifestation of multiple sclerosis. The latter consists of a persisting irregular rippling movement of the facial muscles that usually subsides after a week or two. It supplies the stylopharyngeus muscle and the constrictor muscles of the pharynx. Sensory fibres are carried from the posterior third of the tongue, the ear, the fauces, and the nasopharynx, and chemoreceptor and baroreceptor afferents from the carotid sinus. If it is affected alone, it is very difficult to detect the deficits expected from its anatomical distribution. Lesions usually occur in conjunction with involvement of the vagus and give rise to some dysphagia, impaired pharyngeal sensation, and loss of taste over the posterior third of the tongue. It may be affected in the jugular foramen syndrome, along with the tenth and eleventh nerves, of which glomus tumours or metastatic carcinomas are the commonest causes. The nerve may also be involved in diphtheritic neuropathy, Fisher syndrome, and in cranial polyneuropathy. Glossopharyngeal neuralgia is rare and resembles trigeminal neuralgia but with symptoms in the distribution of the glossopharyngeal nerve. As with trigeminal neuralgia, it is most often encountered in elderly subjects, and the pain may initially be confined to individual branches. Thus, it may be felt deep in the ear, related to the tympanic branch, or in the throat, related to the pharyngeal branches. If that and other antiepileptic drugs fail, surgical treatment, usually section of the nerve, may be required. Nuclear or high vagal lesions, as well as involving the larynx, cause palatal and pharyngeal paralysis. The uvula is pulled up to the opposite side on phonation and pharyngeal sensation is impaired on the affected side. The gag reflex is a reliable indicator if asymmetric, but is bilaterally absent in some normal people. With bilateral paralysis, the palate is paretic leading to nasality of the voice and nasal regurgitation of liquids on attempts at swallowing. Bilateral palatopharyngeal paralysis may be encountered in motor neuron disease, bulbar poliomyelitis, diphtheritic neuropathy, and cranial polyneuropathy. Intrinsic laryngeal paralysis from lesions of the recurrent laryngeal nerve, if unilateral, may be asymptomatic or give rise to hoarseness of the voice. If the superior laryngeal nerve is also involved leading to paralysis of the cricothyroid muscle, the affected cord lies in a paramedian or cadaveric position. The effects of bilateral lesions of the recurrent laryngeal nerves depend upon the degree of approximation of the vocal cords. Lesions of insidious onset give rise to dysphonia and also to stridor on exertion. In partial lesions, close approximation of the cords may result from selective paralysis of the abductor muscles, giving rise to limitation of the airway and sometimes necessitating tracheostomy. With bilateral lesions involving both the recurrent and superior laryngeal nerves, both cords are paralysed and in the cadaveric position. The nerve enters the foramen magnum and briefly joins the cranial portion (functionally part of the vagus nerve) before emerging from the skull through the jugular foramen. The spinal accessory nerve then separates and supplies the sternomastoid and trapezius muscles, the latter also receiving an innervation from the cervical plexus. The nerve may be affected by lesions, often neoplastic, in the region of the jugular foramen, but more commonly it is damaged by injuries to the neck or by operations for the removal of cervical lymph nodes, particularly as it crosses the posterior triangle of the neck. Unilateral paralysis of the sternomastoid usually passes unnoticed by the patient. Paralysis of the trapezius, on the other hand, causes difficulty in lifting the arm above the horizontal, in shrugging the shoulder, and in approximating the scapula to the midline and therefore also in carrying the extended arm backwards. The shoulder droops when the arm is hanging at the side and there is moderate winging of the scapula which is accentuated when the patient attempts to elevate the arm laterally. Motor fibres supply the striated musculature of the palate and pharynx and, through the internal, external, and recurrent laryngeal nerves, the muscles of the larynx. Cutaneous sensory fibres are carried from the external ear and visceral afferent fibres are carried from the pharynx, larynx, trachea, oesophagus, and the thoracic and abdominal viscera. Parasympathetic fibres innervate the parotid gland (through the glossopharyngeal nerve), the heart, and the abdominal viscera. Neurostimulation of the vagus nerve is a treatment for refractory epilepsy mediated by autonomic afferent fibres. The important symptoms of damage to the vagus nerve are those relating to pharyngeal and laryngeal innervation, causing dysphonia and dysarthria; a weak or bovine cough is probably the earliest sign of a mild lesion. The cells of origin in the nucleus ambiguus of the medulla may be damaged in the lateral medullary syndrome, motor neuron disease, and acute bulbar poliomyelitis, leading to dysphagia and dysphonia. Involvement along with the glossopharyngeal nerve in the jugular foramen syndrome has already been mentioned. The recurrent laryngeal nerve may be damaged during operations on the thyroid gland or by tumours within the neck, or within the thorax, usually due to carcinoma of the bronchus. Isolated and unexplained lesions of the recurrent laryngeal nerve are not uncommon. A unilateral lesion of the hypoglossal nerve causes weakness, wasting, and fasciculations of the tongue on the affected side. The protruded tongue may deviate to the affected side if weakness is severe, but this is often misdiagnosed when not truly present. The nerve may be affected by tumours in the region of the anterior condyloid foramen, or by tumours or penetrating injuries in the neck, or by complications of carotid endarterectomy. If damage is the result of a unilateral lower brainstem lesion, it is usually combined with a contralateral hemiplegia. Wasting of the tongue is usually accompanied by fasciculation which is best detected with the tongue at rest. The most common cause is the progressive bulbar palsy variant of motor neuron disease. Many normal people have irregular involuntary twitching or writhing movements of the tongue at rest, but the lack of wasting helps to distinguish this from pathological fasciculations. Once structural lesions are excluded, further investigation depends on suspected cause. Particular conditions affecting the spinal cord Many diseases can affect the spinal cord. Those of particular note include (1) spondylotic myelopathy- the most common cause of progressive myelopathy due to cord compression; (2) multiple sclerosis-may present as an isolated cord syndrome, usually partial rather than complete; (3) transverse myelitis-most commonly affects the thoracic cord; there may be a preceding history of infection, and cerebrospinal fluid analysis may disclose an infective agent; (4) subacute combined degeneration of the cord-demyelination of the posterior and lateral columns due to vitamin B12 deficiency; neurological features may occur in the absence of haematological abnormality; (5) genetic disorders- hereditary spastic paraplegia is usually an autosomal dominant disorder; causative mutations have been described in several genes; (6) vascular disorders-anterior spinal artery occlusion can infarct whole or part of the anterior two- thirds of the cord; (7) syringomyelia; (8) injury/trauma- see Chapter 24. Treatment and prognosis Specific medical and surgical treatments are determined by the particular cause of myelopathy. These may arrest progression, but function that has been lost may not recover fully. Prognosis of acute cord compression is directly related to the time delay between symptom onset and relief of compression. Chronic disability as a consequence of spinal cord disease requires intensive neurorehabilitation. Investigation Intramedullary and extramedullary pathologies may produce distinguishable symptom profiles, but the clinical distinction can only ever be probabilistic. If there is acute onset of myelopathy, and/or structural disease is suspected, imaging of the cord is mandatory, with Introduction Disease within the substance of the spinal cord, intramedullary myelopathy, may result from a wide variety of pathological causes. Clinical features may sometimes give clues to both the pathological nature and anatomical location of disease, but these have been greatly augmented with the development of magnetic resonance imaging studies of the spinal cord. Aetiology and pathogenesis the spinal cord and its addenda may be affected by structural, inflammatory, demyelinating, metabolic, infective, neoplastic, Table 24. Chronic progressive myelopathy should always prompt consideration of a structural lesion such as a tumour, intrinsic or extrinsic, but cervical spondylotic change with osteophyte formation, with or without concurrent intervertebral disc degeneration and prolapse, is the most common cause of progressive cord compression. Hyperacute cord syndromes (evolving over minutes) may result from trauma or vascular pathology, and infective or inflammatory disorders (myelitis) may develop acutely (hours to days) or subacutely (days to weeks), although sometimes present in a chronic progressive fashion (over months).
Generic gyne-lotrimin 100mg with visa
More recent attention has focussed on the promising role of the anti-B-cell monoclonal antibody rituximab pregnancy 0 to 40 weeks discount gyne-lotrimin 100mg, while antitumour necrosis factor- therapy may increase disease activity. Systemic sclerosis Systemic sclerosis results from the excessive deposition of collagen in the skin, and other affected tissues. An association with an acute cerebral vasculopathy is recently reported, but otherwise peripheral nervous system disease predominates, particularly painful trigeminal neuropathy; myopathy, with an elevated creatine phosphokinase also occurs. Mixed connective tissue disease Rheumatoid arthritis An inflammatory peripheral neuropathy occurs in approximately 30% of seropositive rheumatoid cases. A relatively benign mononeuritis is typical, but a more severe and aggressive axonal polyneuropathy or mononeuritis multiplex may be seen when rheumatoid arthritis is accompanied by a vasculitis. More common than either are entrapment neuropathies of conventional distribution, precipitated by synovial swelling. Seronegative arthritides Ankylosing spondylitis Neurological disease in the setting of ankylosing spondylitis usually reflects advanced bony disease; a cauda equina syndrome is wellreported, unexplained, and difficult to treat. Conventionally, the principal neurological manifestations have been held to be peripheral, with descriptions of both a mainly sensory neuropathy and of myositis. Psychiatric abnormalities may occur; spinal cord involvement may take the form of an acute transverse myelitis, a chronic myelopathy, or intraspinal haemorrhage. Occasionally, the features resemble those of multiple sclerosis-and a particular relationship with neuromyelitis optica is emerging. Often, such patients have additional neurological features of peripheral neuropathy or myositis. Psoriasis Included as the third sero-negative arthropathy, the neurology of psoriasis is not extensive. Cord compression from cervical psoriatic spondylosis is described, but reports of a complicating polyneuritis have not been substantiated. Second, there are several primary systemic vasculitides, usually involving the lungs and/or kidneys-for example, polyarteritis and granulomatous polyangiitis-which can also secondarily affect the nervous system. Third, various systemic conditions can include vasculitis-occasionally with neurological involvement-among their complications. These range from rheumatological or connective tissue diseases, to drugs, toxins, and infections. About 50% of patients present with mononeuritis multiplex, the remainder with a more diffuse asymmetrical polyneuropathy or a distal symmetric neuropathy. This grouping carries neither pathological nor therapeutic implications, but may help improve recognition of this condition. Diagnosis and management the diagnosis of cerebral vasculitis involves the exclusion of alternative possibilities (Table 24. Confirming cerebral vasculitis No single simple investigation is universally useful in confirming cerebral vasculitis. Magnetic resonance imaging may disclose ischaemic areas, periventricular white matter lesions, haemorrhagic lesions, and parenchymal or meningeal enhancing areas, but lacks both specificity and sensitivity. Contrast angiography may show segmental (often multifocal) narrowing and areas of localized dilatation or beading, often with areas of occlusion, rarely also with aneurysms. Again, though these changes are not specific, and angiography carries a false-negative rate of up to 50%, and a risk of 10% for transient neurological deficit, and of 1% for permanent deficit. Nuclear imaging of labelled leukocytes, and examination of the ocular vasculature may be useful. Biopsy may reveal an underlying process not otherwise suspected with profound therapeutic implications, such as infective or neoplastic (principally lymphomatous) vasculopathies, but is not a trivial procedure, carrying a risk of serious morbidity estimated at 0. A vasculitic process having been confirmed, the specific defining characteristics of the primary and secondary vasculitides must be painstakingly sought. Neurological vasculitis complicating systemic vasculitides Granulomatous polyangiitis predominantly affects the upper and lower respiratory tracts-nose (often with destructive cartilaginous change causing saddle nose deformity), sinuses, larynx, trachea, and lungs. Microscopic polyangiitis is a multisystem small vessel vasculitis which can involve almost any organ, or may rarely be confined to a single organ. The skin is most commonly involved, usually with purpura or urticaria; the common presence of an allergic precipitant has led historically to the term hypersensitivity vasculitis often being used synonymously in this context; cutaneous leukocytoclastic vasculitis is the currently preferred epithet. Direct effects of the granulomatous process- either by contiguous invasive spread, or from remote metastatic granulomata-represent a mode of neurological involvement unique to granulomatous polyangiitis. Primary invasion of the vascular wall by the infectious agent is, however, the most common precipitant of infection-associated vasculitis. Histoplasma, coccidioides, and aspergillus are among the fungal causes of this picture, usually confined to immune suppressed patients (including individuals with diabetes mellitus). More general bacterial causes of meningeal or cerebral infection- mycobacteria, pneumococci, and H. However, more generalized necrotizing and granulomatous vasculitis, can also occur. The clinical features of cryoglobulinaemia represent the combined consequences of hyperviscosity and of immune complex depositiontriggered vasculitis, particularly in mixed cryoglobulinaemia, when associated with hepatitis C infection. Skin disease, with purpura progressing to necrotic ulceration, and renal and joint involvement are common. Malignancy, lymphomatoid granulomatosis, and malignant angioendothelioma Leukocytoclastic vasculitis (often dermatological) may occur in association with a variety of cancers as a paraneoplastic phenomenon. Lymphomatoid granulomatosis is a lymphomatous disorder centred on the vascular wall, with destructive change and secondary inflammatory infiltration lending the appearance of true vasculitis; the infiltrating neoplastic cell is of T-lymphocyte derivation. Neoplastic or malignant angioendotheliosis is also a rare, nosologically separate disorder, wherein the neoplastic process is intravascular. The neurological features of each disorder are similar, largely representing those of cerebral vasculitic disease; in malignant angioendotheliomatosis, lung involvement is not the rule; characteristic skin manifestations occur. Treatment of cerebral vasculitis Retrospective analyses support the use of cyclophosphamide with steroids in vasculitis. The most compelling evidence of a direct association relates to amphetamines, with clinical and histological evidence of multisystem necrotizing vasculitis. Most strokes occurring with cocaine abuse are associated with arterial spasm, platelet aggregation, severe abrupt hypertension, or migrainous phenomena, not vasculitis, although histologically proven cerebral vasculitis does occur. Infections At least three mechanisms may underlie microbe-related vascular damage-direct invasion, immune complex formation 24. The classical triad of recurrent uveitis with oral and genital aphthous ulceration remains clinically useful, though formal diagnostic criteria have now been proposed and generally adopted. Approximately one-third of patients develop neurological involvement, although this includes the very common occurrence of benign headache. Cerebral venous sinus thrombosis is one of the more specific serious complications; others include sterile meningoencephalitis, encephalopathy, brainstem syndromes, cranial neuropathies, movement disorders, and cortical sensory and motor deficits. Giant cell arteritis Giant cell arteritis, the most common large vessel vasculitis, rarely affects individuals less than 55 years of age. It affects women twice as commonly as men, with an overall annual incidence of some 20 per 100 000 people. Generally, it presents with uni- or bilateral scalp pain, often severe, with exquisite tenderness. Additional symptoms include jaw claudication and polymyalgia rheumatica, with stiffness and aching of the shoulder girdle, worse in the mornings, and occasionally general malaise. The affected temporal artery (-ies) may be thickened and cord-like, often nonpulsatile, and tender.
Order gyne-lotrimin without prescription
As a symptom menopause lubricant order 100 mg gyne-lotrimin visa, for example, the leg stiffness in myotonia congenita lessens as the patient continues to walk. In paramyotonia the reverse is seen, with myotonia increasing with activity-socalled paradoxical myotonia. Some, but by no means all, patients complain that their myotonia is worse in the cold. It is a multisystem disorder that has very important (but sometimes rather neglected) manifestations other than skeletal muscle dysfunction, involving cardiac conduction tissues, smooth muscle, eyes, and the central nervous system. Clinical severity ranges from death in utero to a condition so mild that it may be asymptomatic and with no abnormal physical signs in old age. However, there appears to be a threshold limit for sperm, and males never transmit the very large expansions associated with congenital myotonic dystrophy (see next), which occurs only when the mother is the gene carrier. There is some evidence of meiotic drive, which leads to preferred transmission of the abnormal expanded allele. Clinical features From the previous discussion, it is apparent that there is a continuous distribution of expanded allele size, and a relationship between allele size and disease severity and between allele size and age of onset. Incidence and prevalence figures are unreliable, and probably mostly underestimates, because of the difficulty in identifying asymptomatic individuals. As it is the best known, and illustrates the multifarious manifestations of myotonic dystrophy, the classic form is discussed first. Several rarer or clinically less important associations are also recognized, including reduced fertility, testicular atrophy, insulin resistance (but rarely overt diabetes), retinopathy, eye movement disorder, peripheral neuropathy, disturbed tests of endocrine function, hypotension, pilomatrixomas, and reduced levels of immunoglobulins and complement. There is only a broad correlation between lymphocyte expansion size and clinical severity, in large part because other tissues may have very different expansion sizes compared with lymphocytes. There is somatic mosaicism, so that the expansion is not the same size in different tissues. Neck flexion is weak and in some, but not all, patients there is evident atrophy of the sternomastoid muscles. In the limbs, and in marked contrast to most other myopathic disorders, the weakness is predominantly distal. In the upper limbs there is weakness and wasting of the small hand muscles and of the long wrist, and finger flexor and extensor muscles in the forearm. There is often profound weakness of grip and the patient complains of difficulty with tasks such as wringing out a cloth and removing the lid from a bottle. A simple hand-held dynamometer reveals the extent of the weakness-whereas a normal woman would easily exceed 35 kg, patients of either sex may manage only 1 or 2 kg. In the lower limbs there is weakness of ankle dorsiflexion, presenting as tripping easily and foot-drop. As the disease advances, weakness becomes evident more proximally, but the marked distal predilection remains throughout. There may be evidence of incoordinate uterine contraction in labour but there is little evidence that this is of any clinical significance and most women can deliver a pregnancy normally. The initial manifestation is multicoloured opacities in the subcapsular regions, readily seen on slitlamp examination. In practice, the cataracts are managed as any other cataracts, being operated on when vision is significantly impaired. Early-onset cataracts, even in the absence of any other suggestive features, should always raise the suspicion of myotonic dystrophy. Central nervous system Central nervous system disease is expressed in two main ways. As a group, patients with myotonic dystrophy have a lower intelligence than average, but many mildly affected patients have intelligence within the normal range. There is neuropsychological evidence of specific defects of frontal lobe functioning. The second principal feature is excessive daytime sleepiness, which affects over threequarters of patients, some profoundly. This appears to be a central phenomenon and is only rarely attributable to obstructive sleep apnoea/nocturnal sleep disturbance. Cardiovascular Cardiovascular dysfunction is arguably the most important extramuscular manifestation of myotonic dystrophy and is probably responsible for most of the not infrequently reported cases of sudden death. Tachyarrhythmias also occur, most frequently atrial flutter or fibrillation, but also ventricular arrhythmias, which may be fatal. Rhythm disturbances precipitated by anaesthesia or surgery are common, as are respiratory problems. For these reasons, patients should carry a medical alert bracelet/medallion and, for elective admissions for surgery, be reminded to inform the anaesthetist of their diagnosis. Heart muscle disease, as opposed to disordered cardiac conducting tissues, is not clinically significant and routine echocardiography is not required. Respiratory Recurrent chest infections are common and relate to respiratory muscle weakness and the tendency to aspirate. Respiratory insufficiency may become apparent following anaesthesia, with difficulty in weaning from the ventilator. Chronic hypoventilation and sleep fragmentation may cause excessive daytime sleepiness, but in practice are much less common than the presumed central mechanism already mentioned. Congenital form By definition, this form of myotonic dystrophy is evident at birth, but the spectrum of early-onset myotonic dystrophy is much wider, as noted next. The exclusive (with only very rare exceptions) maternal transmission of congenital myotonic dystrophy has already been discussed. Many fetuses carrying large expansions are aborted spontaneously in early pregnancy and there is a high rate of fetal wastage. In that situation, the diagnosis in the infant is not always immediately apparent, because there are no entirely specific clinical features. There is often a history of polyhydramnios and poor fetal movement in the pregnancy. Respiratory and feeding difficulties may necessitate assisted ventilation or an oxygen tent, and feeding by nasogastric tube. Some die in the neonatal period from respiratory complications, but, somewhat surprisingly, there are few further deaths in the survivors until the late teens and early adult life. There is generalized weakness, including the face-the jaw hangs open and the mouth has a characteristic tented or carp-like (as in fish) appearance. Myotonia is not evident clinically and even electromyographically may not appear for several years. Half survive into the mid-30s, death most commonly resulting from respiratory involvement, but with a proportion of sudden deaths almost certainly due to cardiac conduction defects. Childhood-onset form It is only recently that the specific problems of childhood-onset disease have been recognized. Problems are often first recognized around the start of schooling with evidence of cognitive delay and poor language development. It is typically asymptomatic or oligosymptomatic, and diagnosed during family studies or by an alert ophthalmologist when the patient presents with cataracts. Skeletal muscle disease may be absent, or confined to mild myotonia and weakness restricted to the hands. It is not uncommon to see the parents of a patient with the classic adult form of the disease and not be able to identify the transmitting parent on clinical examination. Importantly, even patients with such minimal symptoms may occasionally develop significant cardiac conduction problems and they should have annual electrocardiograms. A particular concern relates to the genetic phenomenon of anticipation and the potential for an asymptomatic mother, ignorant of the diagnosis, to give birth to a congenitally affected child. When the diagnosis of myotonic dystrophy is established in a family member it is imperative that at-risk relatives are offered screening. Reproductive options include prenatal diagnosis, by chorionic villus sampling, with termination of an affected fetus and, gradually becoming more widely available, preimplantation genetic diagnosis. They and their medical attendants must be aware of the cardiorespiratory complications associated with anaesthesia.
Order gyne-lotrimin no prescription
Stroke often occurs as the first presentation of the disease women's health bendigo phone number order gyne-lotrimin, with a mean age of onset of 36 years. Neurologic symptoms show intrafamilial variation with several unique phenotypes with overlapping features and affected individuals present with infantile hemiparesis, seizures, visual loss, dystonia, strokes, migraine, mental retardation, cognitive impairment, and dementia. Neuroimaging shows diffuse leukoencephalopathy associated with dilated perivascular spaces and microbleeds in several areas, indicating involvement of the cerebral vasculature. Channelopathies and hemiplegic migraine Although not degenerative in nature, these are single gene disorders causing migraine and thus briefly mentioned here. Features of the headache and aura are indistinguishable from the wild-type migraine. Impaired function leads to reduced activity or decreased affinity for K+, leading to impaired clearance of K+ and glutamate from the extracellular space. Several factors are implicated such as a mitochondrial defect may reduce the threshold for migraine attacks. Mitochondrial energy reserves may be diminished in cortical areas, or the trigeminal nerve nucleus or brainstem structures. Exogenous stimuli may create an imbalance between neuronal energy supply and energy consumption, and accelerate the rise in lactate concentration normally occurring with stimulus-induced neuronal activity. Alternatively, the presence of mitochondrial abnormalities in the wall of meningeal blood vessels might explain the sensitivity of such structures to exogenous stimuli. Wilson disease is characterized by the reduced biliary excretion of copper, and reduced incorporation of copper into the serum copper-containing protein ceruloplasmin, resulting in the toxic accumulation of copper in tissues including the liver, kidney, cortex, basal ganglia, and cornea. Incidence is reported at 2 to 4 cases per 100 000 live births and prevalence is estimated at approximately 2 to 3 cases in 100 000 population. Neuropathological studies reveal pigmentation and spongy degeneration of the putamen and to a lesser extent the dentate nuclei, substantia nigra, cerebellar cortex, and thalamic and midbrain nuclei. Approximately 40% of patients, usually children, first present with signs of liver disease including recurrent episodes of jaundice with clinical signs of chronic liver failure and portal hypertension. Neurological signs may herald the onset of the disease in 40% of cases but only after late childhood or adolescence. The first sign may be tremor in one arm or rigidity about the mouth with dysarthria and dysphagia. A masked face and characteristic grin develop, and chewing and swallowing become difficult. Other extrapyramidal signs may appear such as choreoathetosis and in some cases cerebellar ataxia and intention myoclonus. A psychiatric disturbance is the main presenting feature in 20% of patients with depression, anxiety, loss of emotional control, cognitive change, and intellectual decline. Kayser-Fleischer rings may be observed on slit-lamp examination in 50% to 60% of patients with a hepatic presentation of Wilson disease, and 98% of patients with a neurologic presentation. Sunflower cataracts due to copper deposition in the lens may also be observed on slit-lamp examination. High level of clinical suspicion in children presenting without an obvious cause of liver disease and in adults extrapyramidal and/or psychiatric disease must be maintained. Diagnosis is aided by the measurement of serum ceruloplasmin (usually low and less than 15 mg/dl in Wilson disease) and urinary copper (Values are usually greater than 100 g/24 h in symptomatic untreated patients). In a mixed European population, one mutation, H1069Q, accounts for 35 to 45% of disease-causing alleles. Among Asians, 57% of the alleles contain the R778L mutation and, in Russian patients, 40 to 45% have the H714Q and delC2337 mutations. Presence of two of the following criteria is considered diagnostic: positive family history, Kayser-Fleischer rings, Coombsnegative hemolytic anaemia, low total serum copper and ceruloplasmin, elevated hepatic copper, and increased 24-hour urine copper. A combination of Kayser-Fleischer rings and a low serum ceruloplasmin are considered pathognomonic for Wilson disease. In the absence of Kayser-Fleischer rings, an elevated hepatic copper concentration and other biochemical tests helps confirm the diagnosis. The primary goal of therapy is to induce a negative copper balance by inducing urinary copper excretion and preventing copper absorption from the gut. In symptomatic patients, chelating agents such as penicillamine or trientine, which promote urinary copper excretion are mainstay of treatment. Penicillamine is therefore not the treatment of choice as initial therapy to patients with predominant neurologic symptoms and signs. Due to its favourable adverse event profile, Trientine is preferable to penicillamine as first-line treatment. Zinc induces the copper-binding protein metallothionein in the gut mucosa, inhibits intestinal copper absorption and increases excretion in the stool. Lifelong maintenance therapy is requored with zinc or chelation after initial stabilization of symptoms and biochemical abnormalities which takes up to a year. Asymptomatic individuals should receive lifelong maintenance doses of zinc or trientine. Chelation is highly effective in improving hepatic and neuropsychiatric symptoms and signs; however, it may be years to reach maximum improvement in liver function or neuropsychiatric disease. Long-term zinc therapy in a presymptomatic paediatric population improves liver function without adverse effects on growth and development. Liver transplantation may be necessary for decompensated liver disease unresponsive to medical treatment and in patients who present with acute liver failure. Liver transplant may be curative and those who survive the first year generally have a good long-term prognosis. Further investigational therapeutics are focused on gene therapy, gene repair, and hepatocyte transplantation. Supportive therapy includes avoidance of high copper foods such as liver and shellfish indefinitely. Annual 24-hour urinary copper excretion, zinc levels in patients taking zinc, or free copper levels in patients taking chelation therapy, are useful for monitoring. Physical, occupational, and speech therapies can help maximize residual function; counselling the risk of aspiration is important for those with neurological dysfunction. Muscle relaxants and/or botulinum toxin may be useful for symptomatic relief of dystonia. Psychiatric disease needs therapy with antidepressant therapy or antipsychotics as deemed necessary. Vaccination against viral hepatitis A and B is recommended to prevent additional insult to the liver. Memory deficits, agitation, depression, impulsiveness, delusions and hallucinations, and poor judgement are neuropsychiatric features. Over time patients develop hand clumsiness, gait abnormalities, parkinsonism, chorea, dystonia, dysphagia, and tremor, as well as oculomotor disturbances. In juvenile patients, the clinical picture is one of bradykinesia, rigidity, seizures, and dementia. The trinucleotide rereat is translated into a polyglutamine chain and this is associated with accumulation of abnormal protein within th cell. Brain imaging discloses marked flattening of the head of the caudate nucleus and atrophy of the putamen. Tetrabenazine, a central monoamine depleter, and amantadine have both shown improvement in the mean total maximal chorea scores from the Unified Huntington Disease Rating Scale. In its classic adult-onset form it presents with changes in personality and behaviour as well as with involuntary motor movements. Incidence rates are 8 to 18/ 100 000 person-years with a higher prevalence among men than among women and an average age of onset of 60 years. The cardinal clinical features are tremor at rest, slowed movement (bradykinesia), rigidity, and postural instability. Motor block or freezing is particularly disabling, involving a sudden inability to move the feet. Common nonmotor manifestations are autonomic failure, cognitive decline, depression, apathy, hallucinations, and sleep disorders. Younger patients are also at higher risk for levodopa-induced dyskinesias than older patients. Symptoms begin typically after 50 to 80% of dopaminergic neurons in the substantia nigra are no longer functional. The remaining intact nigral neurons may contain intracytoplasmic inclusions (Lewy bodies) composed of aggregates of -synuclein.
