

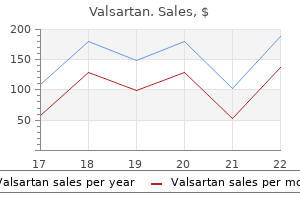
Cheap generic valsartan canada
When the latter are prominent and accompanied by bronchiolitis blood pressure is low buy valsartan cheap, the possibility of exacerbation of an underlying connective tissue disease. Giant cells and scattered small nonnecrotizing granulomas (center) are the only relatively specific markers for lung injury due to methotrexate. Chronic Diffuse Lung Diseases Amiodarone Amiodarone is the drug of choice for the treatment of certain refractory cardiac arrhythmias. Pulmonary toxicity has been reported in 5% to 10% of patients taking this medication. The clinical onset is characterized by slowly progressive dyspnea and dry cough occurring within months of initiating therapy. Approximately one-third of patients experience an acute febrile illness mimicking infectious pneumonia. In children, fibrosis develops in the upper lung zones, with peripheral accentuation. Busulfan Busulfan is an alkylating agent that has been used in the treatment of chronic myelogenous leukemia. Pulmonary toxicity has been reported to occur in 4% of patients,238,311,312 most commonly as acute lung injury. Unfortunately the prognosis for patients with busulfan-induced acute lung disease is poor. Lung toxicity appears to be dose-related, but irradiation or oxygen therapy may predispose the lung to injury. The most common pathologic change seen in amiodarone toxicity is a cellular interstitial pneumonia associated with prominent intraalveolar macrophages whose cytoplasm shows fine vacuolation. Published reports have described the presence of characteristic lamellar cytoplasmic inclusions ultrastructurally. Unfortunately these cytoplasmic and ultrastructural changes are expected with treatment with this drug, so that the mere presence of such changes is not sufficient to warrant a diagnosis of amiodarone toxicity. Targeted Molecular Therapies Progress in the application of newer anticancer therapies using antibodies and small molecules directed at molecular signaling pathways has led to increasing reports of pulmonary toxicity. When intravenous drug use entails injection of crushed analgesic tablets nowadays, microcrystalline cellulose is the commonly identified injurious particle in the lung parenchyma. The physical characteristics and histochemical staining reactions of this material are different than those of talc; these may sometimes be useful distinguishing features. Cornstarch is another filler agent; it appears as a spherical structure with a "Maltese cross" pattern under plane-polarized light. Intravenous foreign material may rarely become encrusted with iron (ferruginated), and sometimes colored tablet coatings (such as blue crospovidone) may be present and quite striking in histopathologic appearance. Emphysema associated with intravenous drug abuse is typically of the panacinar type (similar to that in 1-antitrypsin deficiency) and has most frequently been associated with methylphenidate (Ritalin) abuse. Pulmonary changes may include acute lung injury with airspace organization in the early phase (A) and fibrosis resembling that of usual interstitial pneumonia as a late consequence (B). The mechanism of lung injury is unclear, but some data suggest a hypersensitivity-type reaction, including a high level of helper T cells and a favorable response to corticosteroids. Some of the postulated mechanisms include synergism with cigarette smoke, direct toxic effects of the drug, and induced intravascular leukocyte sequestration causing proteolytic pulmonary injury. Because the foreign material remains in the lung, progression of the clinical and pathologic lesions may occur after discontinuation of intravenous drug use. Recurrence of the changes of intravenous drug abuse have been described rarely in transplanted lung tissue, although such recurrence appears to be a consequence of resumed intravenous drug abuse. A practical approach to lung biopsies with granulomas is presented at the end of this chapter. One complication of intravenous drug abuse is lung fibrosis (A); sometimes this can be massive (B). Practical Pulmonary Pathology Clinical Presentation Sarcoidosis is a systemic disease of uncertain etiology and with frequent lung manifestations. In symptomatic patients, restrictive defects and decreased diffusing capacity are commonly described. Radiologic Findings the clinical staging of sarcoidosis is based on the findings of chest radiography. Five stages of pulmonary sarcoidosis are described, corresponding to the extent of disease. Multinucleated giant cells are characteristically present in the disease, often accompanied by a variety of distinctive cytoplasmic inclusions. The characteristic pathologic lesion of pulmonary sarcoidosis is the nonnecrotizing (immune) granuloma (A), typically occurring within sclerotic fibrosis (B). In sarcoidosis, small granulomas have a tendency to coalesce to form larger nodular lesions, all embedded in refractile eosinophilic collagen. This image is diagnostic of sarcoidosis, but berylliosis should always be included as a diagnostic possibility. Despite this potential for vasocentric growth, pulmonary hypertension is an uncommon complication of sarcoidosis. A variable (but rarely intense) rim of lymphocytic inflammation is typically seen at the periphery of confluent granulomas. A mild inflammatory interstitial infiltrate is said to occur occasionally in pulmonary sarcoidosis, but in practice this is rarely seen. Gilman and coworkers showed that the chance of obtaining a positive result in patients with sarcoidosis increased to 90% when four biopsy specimens were obtained. Special stains for organisms (acid-fast stains and silver stains) should be routinely used when granulomas are identified in lung biopsies to exclude infection, even in the absence of necrosis. In a retrospective study performed by Hsu and colleagues, positive microbiologic cultures were identified in 11% of biopsies in which granulomas were present despite negative special stains of tissue sections. As might be expected, necrosis in granulomas was more frequently associated with culture-positive cases. Differential Diagnosis Granulomatous infection leads the differential diagnosis for sarcoidosis and is the diagnosis of exclusion. Chronic Berylliosis Beryllium, derived from the mineral beryl, is the etiologic agent for berylliosis. Multinucleated giant cells characteristically are present, often accompanied by a variety of distinctive (but not specific) cytoplasmic inclusions: (A) asteroid body; (B) Schaumann body; (C) Schaumann (conchoidal) bodies; (D) Schaumann body in polarized light. In bronchoscopic or transbronchial biopsies, granulomas may be quite dramatic in appearance (A), but sometimes the histopathologic pattern is more subtle. However, recent reports indicate that individuals with low-level exposure, such as residents living near facilities that use beryllium, can also develop berylliosis (for additional discussion, see Chapter 9). The chronic form of berylliosis is indistinguishable from sarcoidosis on histopathologic grounds. Hypersensitivity Pneumonitis (Extrinsic Allergic Alveolitis) Environmental antigens (typically, "organic" protein antigens) are known to produce characteristic inflammatory reactions in the lung in certain predisposed individuals. Similar reactions to ingested antigens associated with some medications can also occur. In general usage, however, hypersensitivity pneumonitis refers to disease occurring as a result of inhalation exposure. The more common antigens implicated in hypersensitivity pneumonitis are presented in Table 8. Clinical Presentation Hypersensitivity pneumonitis can occur as acute or subacute and chronic forms. Like sarcoidosis, chronic berylliosis produces variable fibrosis (A) and distinct granulomas with giant cells (B). There may be a suggestion of more lymphocytic inflammation, but this finding is not sufficiently reliable to be useful diagnostically. Affected individuals experience malaise, dyspnea, dry cough, and occasionally fever and chills. With this symptom complex, the differential diagnosis will include viral infection. Cigarette smoking seems to reduce the risk of developing hypersensitivity, although the mechanism for this protective effect is unknown. Unfortunately the chronic form of hypersensitivity pneumonitis may be progressive, eventuating in death from end-stage lung fibrosis.
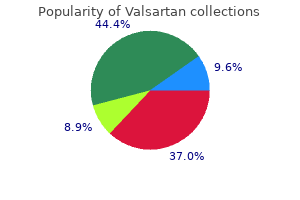
Generic valsartan 160 mg free shipping
Such tumors show scattered cells with varying degrees of degenerative nuclear change blood pressure chart template discount valsartan 40mg fast delivery, including smudgy chromatin and lack of nucleoli (A, B). Complete loss of p16 expression in tumor cells further supports this diagnosis (note retained expression in non-neoplastic vascular/perivascular cells) (B). Like localized intraneural neurofibromas, plexiform tumors often feature the "shredded carrot" pattern of collagen deposition. Of all neurofibromas, the plexiform variant is most prone to undergo malignant transformation, although paraspinal examples of transformation in nonplexiform neurofibromas also show this tendency. Nonetheless, a diagnosis of malignant 346 transformation in a neurofibroma should be made with caution. The simple finding of scattered large cells with marked nuclear pleomorphism and hyperchromasia may or may not be of importance. In most examples, the features of a high-grade malignancy emerge and the diagnosis is straightforward. The ultrastructural features of plexiform tumors and of tactile-like structures have been well studied60,61 and are the same as those discussed previously under "Localized Intraneural Neurofibromas. Plexiform neurofibromas are often resected due to their mass effects or, in superficial lesions, for cosmetic purposes. It has been estimated that as many as 10% of plexiform neurofibromas undergo malignant transformation. Patients whose tumors transform often experience pain and rapid tumor enlargement. Microscopic cross sections of affected fascicles show variable mucin accumulation (A), remnants of the parent fascicle at their centers (B), and strong reactivity on Alcian blue stain (C). Pseudo-meissnerian corpuscles in large numbers may be found between nerve fascicles (A). The lesion is highly cellular, consists of sizable hyperchromatic cells, and is mitotically active (B). Of the few lesions that enter into the differential, most affect skin and superficial soft tissue. Plexiform schwannoma differs in its uniform composition of Schwann cells; the occasional presence of Verocay bodies; relative lack of stromal mucin; lack of a diffuse, extraneural component; the larger size of its cells; and uniform S-100 protein immunopositivity. These include both reactive (traumatic neuroma, interdigital neuroma) and neoplastic processes (perineurioma, neurofibroma, rare malignant peripheral nerve sheath tumors). One is intraneural, and the other, with its variants, is an entirely soft tissue neoplasm. Nonetheless, no association with either form of neurofibromatosis or schwannomatosis is established, although rare exceptions have been reported. General Comments Although at one time considered reactive in nature, intraneural perineuriomas are now recognized as neoplasms. Clinical Manifestations and Localization Intraneural perineuriomas often come to attention in adolescence and early adulthood. Individual fascicles are readily discernible, appearing coarse and being firm in texture. This small bowel lesion shows abrupt transition to transmural involvement by neurofibroma. Histopathology the architecture of intraneural perineurioma is best seen on cross section of the nerve or, preferably, of a single sampled fascicle. Fascicles may not be uniformly affected, some showing early and others well-established changes. As a rule, cytologic atypia is minor; nuclei are elongate and flattened, chromatin is delicate, and mitoses are absent or rare. This colonic tumor shows transmural involvement including submucosal and serosal plexiform elements (A), as well as permeation of the mucosa by tumor cells (B). Reactive intraneural perineurial proliferation is occasionally seen, particularly in the tongue. Such small lesions are submucosal, show dense perineurial collagen deposition, occasional disruption of perineurium, and hyalinized pseudo-onion bulbs. The latter most often affects the median nerve or its digital branches and may be associated with macrodactyly, although other nerves can also be affected. Differential Diagnosis Given the superficial similarity of pseudo-onion bulbs to true (Schwann cell) onion bulbs as seen in hereditary sensorimotor neuropathies (Dejerine-Sottas disease, Charcot-Marie-Tooth disease), these conditions enter into the differential diagnosis. The distinction is made largely on clinical grounds since, in contrast to intraneural perineurioma, both of these disorders are hereditary and generalized. Also in the differential is localized hypertrophic neuropathy, a rare lesion composed Soft Tissue Perineurioma Definition this extraneural soft tissue tumor consists of well-differentiated perineurial cells. General Comments Although this distinctive tumor was once termed "storiform perineurial fibroma," its perineurial nature is now well established. Clinical Manifestations and Localization Unlike intraneural perineurioma, extraneural examples show a slight female predilection and, regardless of site, occur in mid to later adult 351 Practical Surgical Neuropathology shows regressive changes akin to those occurring in schwannomas. These include myxoid degeneration, dystrophic calcification, and even osseous metaplasia. Histopathology Although the tumors have discrete margins, there is no discernible encapsulation. Cytologic features include variation in nuclear shape, ranging from elongate and curved to often disk shaped and twisted. Marked degenerative atypia, as seen in ancient schwannomas and neurofibromas, is infrequent. Cross sections of two fascicles show abundant pseudo-onion bulbs as well as interfascicular involvement (A). Pseudo-onion bulbs show multilayering of perineurial cells around nerve fibers (B). Discrete but devoid of a capsule, they are nodular or ovoid in configuration and range from 0. On cut surface, soft tissue perineuriomas are most often white to tan and hard or rubbery. Clinical Manifestations and Localization Most are solitary, but nearly 10% are multiple. Skin and subcutaneous tissue is mainly affected, particularly of the head, neck, trunk, and extremities. Viscera such as the larynx and gastrointestinal tract are only infrequently involved, multiplicity of lesions being common. Gross Pathology Granular cell tumors usually form solitary nodules less than 3 cm in size. Ultrastructural features include long narrow processes, micropinocytotic vesicles (arrows), and discontinuous basal lamina (arrowheads). Histopathology the tumor cells are disposed in sheets and nests accompanied by fibrous connective tissue. Limited infiltration of adipose tissue is occasionally seen, as is perilesional chronic inflammation. Multinucleation, nuclear pleomorphism, and readily identifiable nucleoli or mitotic figures are uncommon. Overlying skin or squamous mucosa typically shows pseudo-epitheliomatous hyperplasia, which may be associated with alarming atypia and prompt a misdiagnosis of squamous cell carcinoma. Recurrences are exceptional,87 and none has metastasized following such treatment. Granular Cell Tumor Definition and Synonyms Granular cell tumors are distinctive neoplasms with granular eosinophilic cells containing abundant lysosome-rich cytoplasm. Previously utilized names include "granular cell myoblastoma," "granular cell neurofibroma," and "granular cell schwannoma," a reflection of various histogenetic hypotheses. As a tumor group, they share many morphologic, immunohistochemical, and ultrastructural features. The current concept is that granular cell tumors are nerve sheath derived, in keeping with their close association with small to midsize nerves, including rare spinal or cranial examples. In this low magnification image, several nerve fascicles are separated from one another by a fibrolipomatous expansion of the epineurium (B). At higher magnification, a small nerve fascicle displays pseudo-onion bulbs reminiscent of perineurioma (C). Such tumors are smooth contoured, relatively unencapsulated, and lack an accompanying nerve. Ultrastructurally, granular cell tumors are highly distinctive, featuring large numbers of pleomorphic secondary lysosomes varying in type to include autophagosomes, residual bodies, multivesicular bodies, and material resembling ceroid lipofuscin.
Trusted valsartan 160mg
Of chronic epilepsy patients who were treated by neurosurgery and diagnosed with ganglioglioma blood pressure medication used for opiate withdrawal discount valsartan 160 mg visa, 88% were seizure free after 7 years of follow-up. Tumor recurrence in these patients occurs in less than 2% of cases within this interval. Anaplastic gangliogliomas can behave in a locally aggressive fashion, seed the leptomeninges, and lead to death. The large cystic component of these lesions is frequently responsible for their mass effect. Despite the large size of these lesions, there is only mild edema and communication with the ventricular system is rare. At operation, desmoplastic infantile astrocytomas and gangliogliomas are often large, occasionally measuring an average of 10 cm in greatest dimension. Their solid component is usually superficial to the brain, often attached to adjacent dura, gray-white, and rubbery or firm due to the large amounts of collagen. Although these relatively discrete neoplasms do not truly infiltrate adjacent cerebral cortex, tumor and brain may be firmly adherent to one another. Histopathology Histologic examination of desmoplastic infantile astrocytomas and gangliogliomas confirms that they are largely extracerebral in location, with variable, superficial extension of tumor into the brain along Virchow-Robin spaces. The latter typically contain small cells of embryonal or astroglial appearance densely aggregated within a reticulin-free fibrillar matrix. Fairly subtle small polygonal ganglion cells and gemistocytic cells may be seen in both fibrillar and desmoplastic regions. The presence of mature neuronal elements leads to the designation of desmoplastic infantile ganglioglioma rather than astrocytoma. Neuronal cells are most conspicuous in the noncollagenous portions, range considerably in size, and may include ganglion cells of fully differentiated or atypical appearance. Calcifications may be encountered, but lymphocytic infiltrates are less frequent and prominent than classic ganglioglioma. Mitotic activity and necrosis are not usually present and are typically restricted to primitive small cell components. Examples with high mitotic rate, microvascular proliferation, and necrosis have been documented, yet this has not translated into poor prognosis for most of these patients. Several reports have now described such tumors arising in older children, adolescents, and young adults. Such cellular aggregates are also foreign to conventional ganglion cell tumors, although these may demonstrate considerable reticulin and collagen deposition. One desmoplastic infantile ganglioglioma that could not be surgically resected owing to an unusually deep location progressed and caused death. It is characterized by the replacement of the granular cell layer by mature, enlarged, and mildly dysmorphic ganglion cells with subsequent increase in cerebellar size. The spectrum of names attached to the dysplastic gangliocytoma of the cerebellum-Purkinjeoma, gangliomatosis of the cerebellum, granule cell hypertrophy of the cerebellum, and diffuse hypertrophy of the cerebellar cortex-reflects the diversity of opinions regarding its histogenesis. They usually come to clinical attention in the third or fourth decades, although a congenital presentation has been described and patients up to 74 years of age have been affected. Genetics the complete spectrum of genetic alterations present in these tumors has not been defined. In each case, either a normal karyotype or nonclonal abnormalities were described. Clinical Manifestations and Localization the neurologic manifestations are chronic in evolution, with mean intervals of 40 months. In the majority of cases, symptoms include ataxia and seizures, or are related to obstructive hydrocephalus, increased intracranial pressure, or cerebellar injury. Cranial nerve deficits may be observed, with atypical presentations including severe orthostatic hypotension and acute subarachnoid hemorrhage. These include neuronal heterotopias in the white matter, olivary nuclear hypertrophy, hydromyelia, cervical syrinx, vascular malformations, polydactyly, and partial gigantism. In both instances, parallel linear striations are present on the surface of the lesion (tiger stripes) that are nearly pathognomonic and represent affected, abnormally thickened cerebellar folia. The enlarged folia often exhibit surface pallor or yellow-white discoloration reflecting aberrant myelination, may be unusually firm, and frequently have some cavitation of their white matter cores. The abnormal cell processes of the latter further contribute to the enlargement of the folia. These changes range from the modest replacement of only the most superficial cells of the granular layer by ganglion cells to replacement of the entire population. In the most severe cases, the molecular layer also shows increased numbers of atypical neuronal cells. Occasionally a collection of granular and abnormal ganglion cells are seen in the subpial zone of the molecular layer, suggesting an altered neuronal migration of the external granular cell layer during cerebellar development. Another consistent finding is the abnormal myelination of the molecular layer, which results from the myelination of the abnormal neurons that populate the granule cell layer, extending axons into the molecular zone. Myelinated axons run in parallel to the pial surface in deeper layers and perpendicular to it more superficially. Other histologic findings include a reduction in the number of Purkinje cells, microcalcifications, large bizarre neurons, dense capillary networks, and vacuolization of the cerebellar white matter. These conventional ganglion cell tumors, however, are distinct from the dysplastic gangliocytoma of the cerebellum in their imaging and histopathology. Histologically, conventional ganglion cell tumors show a more disordered growth of the ganglionic cell component and there is usually a lymphocytic infiltrate noted. Patients with Cowden syndrome should be screened for dysplastic gangliocytomas of the cerebellum, which may be discovered incidentally. Conversely, dysplastic gangliocytomas of the cerebellum may predate the appearance of other manifestations of Cowden syndrome; therefore, these patients should be screened for possible Cowden disease. Central neurocytomas are typically located supratentorially in the lateral ventricles and/or the third ventricle. The anterior portion of one lateral ventricle is the most frequent site (50%), followed by the combined involvement of the lateral and third ventricles (15%) and the involvement of both lateral ventricles (13%). Occasionally, tumors with histopathologic, immunohistochemical, and biologic features similar to central neurocytoma may be found outside the ventricles, within the cerebral hemispheric parenchyma or other scattered sites throughout the neuroaxis. Surgical resection is curative in the majority of cases, but local recurrence also occurs in a substantial percentage of cases with long-term follow-up. Many have suggested that recurrence of these dysplastic gangliocytomas favors a neoplastic rather than hamartomatous designation for these lesions. It has a characteristic location within the lateral ventricles in the region of the foramen of Monro. Prior to this report, most of these lesions were misinterpreted as intraventricular oligodendrogliomas or ependymomas. A neurocytic component may also be encountered in other rare glioneuronal tumors, such as the papillary glioneuronal tumor and the rosette-forming glioneuronal tumor; these entities are discussed separately. Hydrocephalus due to obstruction of the foramen of Monro may be present in central neurocytoma. In the case of central neurocytoma, they often adhere to the ventricular lining or septum pellucidum. Histopathology the diagnostic features of central neurocytoma are distinctive on histologic exam, but verification usually requires immunohistochemical evidence of neuronal differentiation. Tumor cells grow either in a solid pattern or in linear arrays and often cluster around small blood vessels simulating the perivascular pseudorosettes of ependymoma, but containing delicate neuropil rather than coarse glial processes. According to a comprehensive literature review of central neurocytoma, the majority were found in young adults; the mean age at clinical presentation was 29 years, 46% being diagnosed in the third decade with approximately two-thirds presenting between the ages of 20 and 40 years. Localization and Clinical Manifestations the majority of patients present with signs and symptoms of increased intracranial pressure, such as headache, nausea, and vomiting, rather than with a distinct neurologic deficit. Such neuropil arises from delicate tumor cell processes and is distinctive from the coarser fibrillarity of astrocytic and ependymal neoplasms. In some lesions, clear cells predominate, mimicking the pattern of oligodendroglioma.
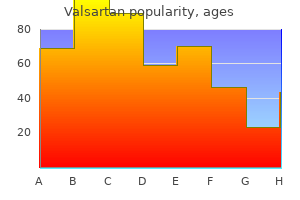
Buy cheap valsartan 40mg on line
Arachnoid cysts vary from thin (A) to thickened (B) hypertension treatment algorithm purchase 80 mg valsartan, with an inner layer of loose arachnoidal tissue and an outer layer of collagen. Meningothelial cells are often flattened (A), but may show clusters of more typical cap cells (B; arrows). In the second example, the thickening may be partially due to prior hemorrhage, as evidenced by collections of hemosiderin-laden macrophages. Breast adenocarcinoma metastatic to epidural cervical spine meningioma: case report and review of the literature. Intrameningioma metastasis as first clinical manifestation of occult primary breast carcinoma. Melanoma metastatic to central neurocytoma: a novel case of tumor-to-tumor metastasis. Metastasis of renal cell carcinoma to central nervous system hemangioblastoma in two patients with von Hippel-Lindau disease. Immunohistochemical differentiation of hemangioblastoma from metastatic clear cell renal carcinoma: an update. Inhibin alpha distinguishes hemangioblastoma from clear cell renal cell carcinoma. Immunohistochemical analysis of metastatic neoplasms of the central nervous system. Application of immunohistochemistry to the diagnosis of primary and metastatic carcinoma to the lung. Immunohistochemical profile and chromosomal imbalances in papillary tumours of the pineal region. Spontaneous rupture of spinal dermoid cyst with disseminated lipid droplets in central canal and ventricles. Congenital intracranial frontotemporal dermoid cyst presenting as a cutaneous fistula. Malignant transformation of intra-cranial epithelial cysts: systematic article review. Recurrence of a neurenteric cyst with malignant transformation in the foramen magnum after total resection. Well-differentiated papillary adenocarcinoma arising in a supratentorial enterogenous cyst: case report. Transcortical-transventricular approach in colloid cysts of the third ventricle: surgical experience with 26 cases. Organic and psychogenic factors leading to executive dysfunctions in a patient suffering from surgery of a colloid cyst of the Foramen of Monro. Colloid cysts of the third ventricle with fatal outcome: a report of two cases and review of the literature. As with any brain tumor, clinical features are related to the brain regions involved. Extensive brain invasion with single tumor cells has been designated lymphomatosis cerebri, being similar to the extensive diffuse invasion patterns of glioma cells in gliomatosis cerebri. Necrotic areas are often incomplete and less well demarcated than in glioblastomas. There is a marked reaction of non-neoplastic cells, including numerous macrophages (in solid areas of high tumor cell density) or microglial cells (in areas with more diffuse invasion), as well as reactive T cells and astrocytes, the latter of which may have pleomorphic or even multiple nuclei. Features that favor the diagnosis of lymphoma over malignant glioma include the presence of abundant apoptotic nuclei, intermixed small (reactive) lymphocytes, stellate astrocytes of typical "reactive" morphology, and the absence of microvascular proliferation and palisading necrosis that are often seen in glioblastomas. This latter scenario is particularly common, especially if the patient was treated with steroids prior to biopsy. Frozen section diagnosis is often difficult due to increased pleomorphism and artifacts simulating background fibrillarity (A). However, intraoperative smears show superior cytologic preservation with classic features such as lack of cohesion, scant cytoplasm, and nucleolar prominence (B). This form of lymphoma is characterized by angionecrosis and perivascular infiltrates resembling poorly formed granulomas or lymphohistiocytic nodules (A, H & E). Patients are usually immunosuppressed, although this may be detected only after careful clinical or laboratory analysis. Histologic distinction from other cases of immunodeficiency-associated lymphoma is vague and subjective. In fact, general pathologists who are not so familiar with the typical angiocentric pattern of any lymphoid lesion within the brain tend to be more inclined to making the diagnosis of intracerebral lymphomatoid 410 granulomatosis as compared to neuropathologists. A defect in homing receptors preventing extravasation of tumor cells has been postulated. Even today, the diagnosis is often only made at autopsy, since the clinical presentation is often clinically confusing. However, it is occasionally encountered at the time of biopsy, and the diagnosis is easily missed without special attention to the intravascular cellular composition. There is variable plasmacytic differentiation, and when prominent, the differential diagnosis may include IgG4-related disease. In contrast to other types of intracranial lymphoma, lymphoid follicles may be encountered, but are often immature or poorly formed. Nonetheless, rarity and diagnostic problems have precluded systematic neuropathologic and molecular analyses. Intracranial plasmacytoma may be localized to the meninges, brain parenchyma, cerebral ventricles, or skull base22; in most cases, it is thought to represent intracranial spread from adjacent bone. Diagnosis may be very difficult in cases dominated by reactive histiocytes (lymphohistiocytic pattern), small tumor cells, or by multinucleated tumor cells. Occlusion of central nervous system vessels by tumor cells leads to multiple hemorrhagic infarcts (A, gross). Cytologically, the tumor cells are small to midsize lymphocytes with pale cytoplasm (C, H & E). Myeloid sarcoma may arise de novo, may precede or coincide with acute myeloid leukemia, or may represent acute blastic transformation of myelodysplastic syndromes. Features of promyelocytic, neutrophilic, myelomonocytic, monoblastic, erythroblastic, or megakaryoblastic differentiation may be present or absent. Other Brain Tumors Histologically Mimicking Lymphoma Lymphomas with a cohesive pattern, in particular large-cell anaplastic lymphomas, may resemble metastatic carcinoma. Similarly, the infiltrative component can mimic diffuse gliomas when the angiocentric pattern is not prominent, especially if the nuclear cytology is poorly preserved. This phenomenon is even more pronounced in the rare cases of lymphomatosis cerebri, in which the infiltrative pattern is essentially identical to that encountered in gliomatosis cerebri. Secondary involvement of the central nervous system by hematopoietic malignancies often involves the dura or leptomeninges (or both) (A, B, gross images of meningeal acute lymphoblastic lymphoma; C, H & E of secondary leptomeningeal lymphoma). Cerebrospinal fluid cytology is often a useful diagnostic tool in such cases (D, lymphoma on Pappenheim stain). Histologic analysis in these cases reveals numerous apoptotic nuclei, patchy and incomplete rather than confluent and complete demyelination, and a fuzzy rather than sharply demarcated lesion edge. Pathogenetically, malignant transformation of a chronic inflammatory process, demyelination due to antimyelin antibodies secreted by lymphoma cells, or even sampling error have been discussed as possible explanations. Histologically, sentinel lesions show variable demyelination, T cell infiltrates, B cells, plasma cells, numerous macrophages, astrogliosis, and relatively well-preserved axons, but no neoplastic B cells. However, biopsy specimens showing only a few lymphoma cells within reactive brain tissue or active demyelinating lesions with proliferating perivascular B cells may pose problems. However, atypical imaging features have been described, which may confound the diagnostic process, including a solitary large lesion, size greater than 2 cm, associated mass effect, perilesional edema, or the presence of ring enhancement. A large series of 168 tumefactive biopsy cases revealed typical histologic features of inflammatory demyelination, although 18% of cases had originally been misinterpreted, most often as glioma and occasionally as lymphoma. A variety of other demyelinating disorders showing a variable inflammatory component usually do not pose a problem in the differential diagnosis with lymphoma when classic histologic, radiologic, or clinical features are found. Encephalitis and Other Inflammatory Conditions Viral encephalitis may rarely present with multifocal mass lesions radiologically resembling lymphoma. Cellular perivascular T cell infiltrates of low to moderate proliferative activity, absence of polymorphic tumor cells, occasional microglial nodules, and the presence of neuronophagia argue against lymphoma.
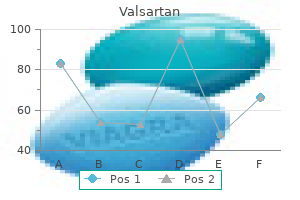
Purchase 40 mg valsartan free shipping
Ependymoma with extensive lipidization mimicking adipose tissue: a report of five cases pulse pressure 45 cheap valsartan 40 mg on line. Ependymoma with neuropil-like islands: a case report with diagnostic and histogenetic implications. Signet-ring cell ependymoma: case report with implications for pathogenesis and differential diagnosis. Oncocytic ependymoma: a new morphological variant of high-grade ependymal neoplasm composed of mitochondrion-rich epithelioid cells. Rare histological variants in ependymomas: histopathological analysis of 13 cases. Eosinophilic inclusions in ependymoma represent microlumina: a light and electron microscopic study. Intraoperative diagnosis of tanycytic ependymoma: pitfalls and differential diagnosis. Clear cell ependymoma: a mimic of oligodendroglioma: clinicopathologic and ultrastructural considerations. Supratentorial clear cell ependymomas with branching capillaries demonstrate characteristic clinicopathological features and pathological activation of nuclear factor-kappaB signaling. Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. Rapid spontaneous malignant progression of supratentorial tanycytic ependymoma with sarcomatous features-"ependymosarcoma". Ependymal tumors with sarcomatous change ("ependymosarcoma"): a clinicopathologic and molecular cytogenetic study. Monomorphous angiocentric glioma: a distinctive epileptogenic neoplasm with features of infiltrating astrocytoma and ependymoma. An immunohistochemical comparison of chordoma with renal cell carcinoma, colorectal adenocarcinoma, and myxopapillary ependymoma: a potential diagnostic dilemma in the diminutive biopsy. Ependymomas with neuronal differentiation: a morphologic and immunohistochemical spectrum. Immunohistochemical study of central neurocytoma, subependymoma, and subependymal giant cell astrocytoma. Microtubular aggregates within rough endoplasmic reticulum in myxopapillary ependymoma of the filum terminale. Expression profiling of ependymomas unravels localization and tumor grade-specific tumorigenesis. Genomic characterization of ependymomas reveals 6q loss as the most common aberration. Low frequency of chromosomal inbalances in anaplastic ependymomas as detected by comparative genomic hybridization. Gain of 1q and loss of 22 are the most common changes detected by comparative genomic hybridization in paediatric ependymoma. Identification of gains on 1q and epidermal growth factor receptor overexpression as independent prognostic markers in intracranial ependymoma. Genomic imbalances in pediatric intracranial ependymomas define clinically relevant groups. Candidate genes on chromosome 9q33-34 involved in the progression of childhood ependymomas. Neuronal differentiation distinguishes supratentorial and infratentorial childhood ependymomas. Molecular sub-group-specific immunophenotypic changes are associated with outcome in recurrent posterior fossa ependymoma. Supratentorial and spinal pediatric ependymomas display a hypermethylated phenotype which includes the loss of tumor suppressor genes involved in the control of cell growth and death. Analysis of chromosome 7 in adult and pediatric ependymomas using chromogenic in situ hybridzation. Predictors of survival among pediatric and adult ependymoma cases: a study using Surveillance, Epidemiology, and End Results data from 1973 to 2007. Distinct disease-risk groups in pediatric supratentorial and posterior fossa ependymomas. Pediatric infratentorial ependymoma: prognostic significance of anaplastic histology. Clinical course and progression-free survival of adult intracranial and spinal ependymoma patients. Multicentric French study on adult intracranial ependymomas: prognostic factors analysis and therapeutic considerations from a cohort of 152 patients. Supratentorial ependymomas: prognostic factors and outcome analysis in a retrospective series of 46 adult patients. Prognostic value of tumor microinvasion and metalloproteinases expression in intracranial pediatric ependymomas. Pediatric intracranial ependymoma: the roles of surgery, radiation and chemotherapy. Region specific differences of claudin-5 expression in pediatric intracranial ependymomas: potential prognostic role in supratentorial cases. Treatment and survival of supratentorial and posterior fossa ependymomas in adults. Supratentorial hemispheric ependymomas: an analysis of 109 adults for survival and prognostic factors. Optimising treatment strategies in spinal ependymoma based on 20 years of experience at a single centre. Adjuvant radiotherapy delays recurrence following subtotal resection of spinal cord ependymomas. Analysis of the prognostic significance of selected morphological and immunohistochemical markers in ependymomas, with literature review. Childhood intracranial ependymoma: twenty-year experience from a single institution. Post-operative radiation improves survival in children younger than 3 years with intracranial ependymoma. Tandem high-dose chemotherapy and autologous stem cell transplantation for anaplastic ependymoma in children younger than 3 years of age. Treatment and outcome of children with relapsed ependymoma: a multi-institutional retrospective analysis. Survival benefit for pediatric patients with recurrent ependymoma treated with reirradiation. Extraneural ependymoma: distant bone, lung, liver, and lymph node metastases following bevacizumab. The histologic grade is a main prognostic factor for patients with intracranial ependymomas treated in the microneurosurgical era: an analysis of 258 patients. Pediatric intracranial ependymomas: prognostic relevance of histological, immunohistochemical, and flow cytometric factors. Ki78 index in intracranial ependymoma: a promising histopathological candidate biomarker. Spinal myxopapillary ependymoma outcomes in patients treated with surgery and radiotherapy at M. Spinal cord ependymomas and myxopapillary ependymomas in the first 2 decades of life: a clinicopathological and immunohistochemical characterization of 19 cases. Outcome predictors in the management of spinal myxopapillary ependymoma: an integrative survival analysis. Epidermal growth factor receptor overexpression is common and not correlated to gene copy number in ependymoma. Nucleolin overexpression is associated with an unfavorable outcome for ependymoma: a multifactorial analysis of 176 patients. The role of resection alone in select children with intracranial ependymoma: the Canadian Pediatric Brain Tumour Consortium experience. Myxopapillary ependymoma in children: a study of 11 cases and a comparison with the adult experience. Telomerase inhibition abolishes the tumorigenicity of pediatric ependymoma tumor-initiating cells. The transcription factor evi-1 is overexpressed, promotes proliferation, and is prognostically unfavorable in infratentorial ependymomas. Study of stem cell marker nestin and its correlation with vascular endothelial growth factor and microvascular density in ependymomas.
Gelsemii Rhizoma (Gelsemium). Valsartan.
- Are there safety concerns?
- How does Gelsemium work?
- What is Gelsemium?
- Asthma, pain due to migraine headaches, pain due to a condition of facial nerves called trigeminal neuralgia, and other uses.
- Dosing considerations for Gelsemium.
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96464
Cheap 160mg valsartan overnight delivery
Pulmonary blastoma with germ cell (yolk sac) differentiation: report of two cases blood pressure stroke valsartan 80mg for sale. Placenta-like alkaline phosphatase reactivity in human tumors: an immunohistochemical study of 520 cases. The use of electron microscopy and immunohistochemistry in the diagnosis and understanding of lung neoplasms. Ultrastructural and immunohistochemical features of common lung tumors: an overview. Cytologic, histologic, and electron microscopic correlations in poorly-differentiated primary lung carcinoma: a study of 43 cases. Appraisal of the comparative utility of immunohistochemistry and electron microscopy in the diagnosis of childhood round cell tumors. A realistic approach to the use of electron microscopy and other ancillary diagnostic techniques in surgical pathology. Electron microscopy in an oncologic institution: diagnostic usefulness in surgical pathology. Uses and contributions of diagnostic electron microscopy in surgical pathology: a study of 20 Veterans Administration hospitals. An ultrastructural comparison of mesotheliomas with adenocarcinomas of the lung and breast. An ultrastructural comparison of mesotheliomas and adenocarcinoma of the ovary and endometrium. Genetics of epithelial tumors of the renal parenchyma in adults and renal cell carcinoma in children. A molecular cytogenetic study of chromosome 3 rearrangements in small cell lung cancer: consistent involvement of chromosome band 3q13. Establishing germ cell origin of undifferentiated tumors by identifying gains of 12p material using comparative genomic hybridization analysis of paraffin-embedded samples. Sensitivity and cost-effectiveness of fine needle aspiration with immunocytochemistry in the evaluation of patients with a pulmonary malignancy and a history of cancer. Clinical perception of the utility of sputa and other tests in patients with lung masses. Treatment and prognosis of metastatic carcinoma of unknown primary: analysis of 100 patients. Conceptually, all malignancies that metastasize to the lungs comprise a single clone of neoplastic cells. The rate at which primary tumors develop the ability to metastasize varies substantially. Tumor-related thrombotic microangiopathy can produce pulmonary hypertension and cor pulmonale. The presence of a solitary intraparenchymal nodule does not exclude the possibility of metastasis. The most common source of a solitary metastatic tumor in the lung that measures more than 5 cm in diameter is the: A. The accuracy of clinicopathologic sampling methods that are used to identify intrapulmonary metastases: A. Is less than 50% with regard to computed tomography-guided transthoracic fine-needle aspiration D. Which of the following sources of information are useful in determining the nature and source of a solitary tumor in the lung Intrapulmonary adenocarcinomas with a papillary appearance commonly originate in the: A. Fine-needle aspiration biopsies that are aimed at sampling lesions in the basal lower lobe of the right lung can yield a misdiagnosis of well-differentiated oncocytic adenocarcinoma because of sampling the: A. Metastatic carcinomas in the lung that may show obvious central necrosis include: A. The single most important characteristic of gene chip analyses of metastatic tumors in the lung is the: A. Which of the following cell lineages may pertain to small cell metastatic tumors in the lung Gene profiling to separate primary from secondary squamous carcinomas in the lung is associated with an accuracy of: A. The central form of hamartoma is encountered in association with large bronchi, as an endoluminal polypoid protuberance covered by intact mucosa. Most of these lesions contain predominantly cartilaginous tissue and are therefore firm to hard, relatively homogeneous, and translucent when transected. These elements are usually represented by hyaline cartilage, but fibrous tissue, smooth muscle, adipocytic components, and bone18 may be seen. Those masses lacking chondroid elements have sometimes been diagnosed as intrabronchial lipoma, myxoma, leiomyoma, or fibroadenoma. Entrapped alveolar epithelium may also undergo cuboidal or low-columnar 19 There is a limited group of pulmonary lesions that one can classify as pseudoneoplastic, but their conditions constitute a significant aggregation in absolute numbers. Other clinical pseudotumors such as amyloidoma and rounded atelectasis1 are confused with neoplasms only by nonpathologists and are not included for discussion here. Indeed, the last of these changes can be prominent and bear a resemblance to placental tissue,19 a feature referred to as placental transmogrification. Chondroid hamartoma as sampled by fine-needle aspiration biopsy, with bland spindle cells surrounded by myxoid stroma (B). Other foci show bland cuboidal or low-columnar epithelial cells (C) and fragments of cartilage (D). True chondromas of the lung tend to be multiple and composed only of cartilage, which is smoothly circumscribed. Simple excision of the lesions is typically performed today, especially with the availability of thoracoscopic video-assisted surgical techniques. There is also likely overlap between some of these lesions and those described in association with IgG4 sclerosing disease (see subsequent discussion). This lesion shows no sex predilection and occurs over a broad age range, from 1 to 77 years, with a mean of 27 to 50 years. Those that contain numerous inflammatory cells are tan-white and fleshy, and those with a predominance of mesenchymal tissue are gray and firm. Fibroblastic proliferation is admixed with fibrinoinflammatory exudate in alveoli, alveolar ducts, and bronchioles. Neutrophils are sometimes interspersed with the lymphocytes and plasma cells, and they may form intralesional microabscesses that result in small areas of cavitation. Multinucleated cells of the Touton type are sometimes present, as are foci of dystrophic calcification, osseous metaplasia, or myxomatous change. Prominently dilated lymphatic spaces, containing histiocytes that show emperipolesis of lymphocytes, may also be observed. Usually, both kappa and lambda light chain immunoglobulins are detectable immunohistologically in the plasma cells, indicating a polytypic population51; lymphocyte subset markers similarly show an admixture of B cells and T cells. The premise that some cases represent a peculiar form of localized pneumonia has support from a history of a previous febrile illness with respiratory complaints in up to 40% of cases. In their hands, p53 was restricted to malignant lesions, albeit with less than absolute sensitivity for such tumors. Spontaneous resolution has also been documented, and a few lesions have shrunk after small incisional biopsy or administration of systemic corticosteroids or irradiation. Mycobacterial Spindle-Cell Pseudotumor Spindle-cell pseudotumors that are reactions to mycobacterial infection have been documented in several organ sites in immunosuppressed patients. Microscopically, the lesions comprise aggregates of spindle cells with a fascicular growth pattern and without significant atypia or mitoses. Scattered lymphocytes and plasma cells may be present, but overt granulomas are lacking. The cytoplasm of the spindle cells is "foamy" but may contain hemosiderin focally. The lesional cells are immunoreactive for lysozyme, with no labeling for S100 protein, keratin, actin, desmin, or von Willebrand factor. That is, a reproducible cross-reaction has been seen with mycobacterial antigens using certain desmin antibodies,90 spuriously suggesting the presence of a myogenous proliferation.
Syndromes
- Above normal level of the hormone insulin in the blood
- Standing up after sitting for awhile
- The type of infection
- Hematoma (blood accumulating under the skin)
- Tremor
- Bleeding in the brain
- Drugs such as amantadine and tetrabenazine are used to try to control extra movements.
Cheap valsartan american express
In carcinoid tumors with abundant mucinous stroma blood pressure medicine generic valsartan 40mg otc, metastatic mucinous carcinomas with uniform cells, as in breast carcinomas, and goblet cell gastrointestinal carcinoids are in the differential diagnosis. Treatment and Prognosis Prognosis is favorable in early-stage disease,161 but outcome is less favorable than typical carcinoids with advanced disease. Although limited resection may be sufficient for typical carcinoids, anatomic resections/lobectomy with nodal dissection have been recommended for atypical carcinoids. In advanced disease, resection of liver metastasis may be considered as in typical carcinoids if disease distribution is otherwise favorable. Given the more aggressive behavior and increased recurrence rate,162 it might be expected that the failure rate of this approach would be high. One series reported curative resection in 5 of 29 patients and a survival of 89% with long-term follow-up. This latter finding, however, may be related to stage or patient selection rather than the specific therapy itself. The cytoplasm of the cells is visible, and the nuclei appear spaced apart as a result, with more abundant cytoplasm in cells away from the periphery. In contrast, features of small cell carcinoma include finer chromatin and nuclear molding. Cellular size, presence of cytoplasm with coarser chromatin pattern, identifiable nucleoli, apoptosis, mitoses, and necrosis are all features seen cytologically. However, when examined, 85% have at least two markers, and 68% have all three positive. However, to date, targeting these pathways in the absence of mutation has not been effective. Staining in small cell carcinoma is often weaker and has a dot-like staining pattern. This can be helpful in small specimens when architecture is limited and cellular crush artifact is prominent. Therefore on small samples, any evidence of small cell carcinoma warrants a small cell designation because combined tumors are considered small cell carcinoma, combined type. It has been reported more often in adenocarcinoma than squamous carcinoma in some series but not others. Loss of 3p, 5q, 11q, 13q, and 5p gain, but these were common to high-grade neuroendocrine carcinoma. Its recognition as an entity dates back to 1926,239 with clearer histopathologic criteria established by Azzopardi in 1959. Although rare, these various paraneoplastic syndromes represent unusual presenting symptoms and disease associations that warrant clinical investigation for malignancy. Unusual metastatic sites can also be a manifestation of this disease, including involvement of the eye with choroidal and uveal metastasis262 and transplacental transfer. This hazard ratio is closely linked to the number of cigarettes smoked per day but does increase with years of smoking. In men, there is a slight increase in risk based on early age of smoking initiation. After smoking cessation, a marked decrease in risk occurs after 5 years, which becomes markedly reduced after 25 years. Only a small percentage of small cell carcinomas are diagnosed in never smokers (<3%). Industries with a risk of small cell carcinoma include steel industries and iron foundries, as well as concrete pouring and motor vehicle parts manufacturing. Radiation exposure in uranium miners may lead to a greater proportion of squamous and small cell carcinoma. This can include chest pain, hemoptysis, and hoarseness, as well as constitutional symptoms of malaise, anorexia, and weight loss. In some instances, compression of the superior vena cava can result in superior vena cava syndrome, with facial edema and reduction of venous drainage of the head and neck. Metastatic disease can present as bone pain or neurologic symptoms from brain metastasis. This is an adverse prognostic feature and needs to be corrected during chemotherapy. Sensory neuropathy and encephalomyelitis are more common in men254 and can be associated with cerebellar symptoms. Rare syndromes, such as opsoclonus-myoclonus syndrome characterized by rapid eye movements and muscle twitching, have also been reported. Invasion of mediastinal structures, compression of the superior vena cava, and bronchopleural fistula formation can be seen. In resected small cell carcinoma, gross features consist of surprising circumscription and some lobulation. Apoptotic bodies and nuclear karyorrhexis are more visible on hematoxylin and eosin (H&E) sections. In the absence of mitoses or apoptotic or karyorrhectic debris, the diagnosis of small cell should be questioned. Although the name implies that the cells should be small (and defined as less than the diameter of three resting lymphocytes), the cell size does depend on the type of fixation and specimen type. Uncrushed resected small cell carcinoma may be composed of cells that appear larger than three resting lymphocytes, but the other features-high nuclear-to-cytoplasmic ratio and nuclear chromatin pattern-are more diagnostic than cell size alone. Organoid and trabecular patterns are described as are rosettes and solid sheet-like areas (eSlide 14. Apoptosis is always present, and mitotic activity is high, which eliminates an atypical carcinoid as a possibility. Large cell neuroendocrine patterns are frequently encountered, in as many as one-third of cases, but exceeding 10% of the tumor in only 16%. Despite its utility, the diagnosis of small cell carcinoma by cytology was underutilized (only 20%) in various treatment settings. Prior terminologies of mixed subtype or intermediate cell type have been abandoned because they were found not to be clinically relevant. Cytokeratin cocktails of low- and high-molecular-weight keratins are positive in a large percentage of small cell carcinomas, in some series nearly 100%. However, a subset of small cell carcinomas will be negative for neuroendocrine markers, and while this could raise a consideration of other diagnoses, it should not prevent the rendering of a small cell carcinoma diagnosis. Crowded, cellular cluster with high nuclear-to-cytoplasmic ratio, apoptosis, and Azzopardi effect. Differential Diagnosis In the evaluation of small cell carcinoma, several diagnoses can be considered and their likelihood influenced by patient age, smoking history, clinical presentation, and quality of the sample. In small samples, frequent crush artifact leads to limited morphologic assessment. Some high-grade carcinomas, such as nonkeratinizing subtypes of squamous carcinoma including basaloid carcinoma, may be a mimicker. As previously discussed, crushed samples of carcinoid and especially atypical carcinoid should be considered, and some advocate Ki-67 in all cases in which morphology is limited. Extrapulmonary high-grade neuroendocrine tumors, such as Merkel cell carcinoma or thyroid medullary carcinoma, may enter the differential diagnosis as well. Given the frequency of a large cell neuroendocrine component in small cell carcinoma, this is a frequent problem. However, interobserver agreement in these two tumor types is only fair, and therefore careful adherence to criteria is essential. Of surgically resected cases, patients with early-stage disease (Stage 1 and Stage 2) have 5-year survival rates that decrease from 56% in Stage 1A and Stage 1B to 40% in Stage 2. Despite these low 5-year survival numbers, this does indicate a potential cure rate in early-stage disease. For early-stage patients, surgical resection may be an option when feasible,289 but surgical therapy alone is not curative. In the absence of nodal disease, the combination of chemotherapy and thoracic radiation rather than chemotherapy alone remains debated. There does not appear to be a benefit for surgery after chemoradiation in these patients. These are usually mitotically active tumors, but a strict cutoff is not established as it is for other neuroendocrine tumors.
Purchase valsartan on line
This example consists of multiple nodules (A) pulse pressure 50 generic 160 mg valsartan with mastercard, the surface aspect of which shows splaying of the constituent cells (B). Most involve skin and occasionally superficial subcutaneous tissue of the face rather than deeper soft tissue. The Schwannoma Schwannomas are among the most common of peripheral nerve neoplasms. Based on differing clinical, gross, and histologic features, three major forms are recognized10: conventional schwannoma, cellular schwannoma, and plexiform schwannoma. These relatively cellular proliferations (A) consist of S-100 protein reactive Schwann cells (B), usually outnumbering neurofilament positive axons (C). This somewhat polypoid example (A) consists of nerve with stromal mucin (B) and a full complement of Schwann sheaths (C, S-100 protein stain) and axons. Nonetheless, the origin of the tumor 330 from a nerve is often demonstrable, particularly if it is carefully sought, and its finding often suggests a nerve sheath tumor rather than another soft tissue neoplasm. With respect to the spinal cord, schwannomas are typically intradural extramedullary masses, often forming dumbbell-shaped tumors with a narrowing where they traverse the spinal foramen. Other cranial nerves, such as the trigeminal and facial nerves, are also occasionally the source of schwannomas, but much less frequently than the vestibular nerve. Clinicopathologic features of schwannomas are further discussed in the next section according to histologic subtype. This most common form of schwannoma generally presents in adults with a peak incidence between the third and sixth decades, there being no gender prevalence. Sites commonly affected include the head and neck, flexor surfaces of the extremities, and spinal as well as cranial nerves, most often of sensory type. In most cases, gross inspection of a schwannoma provides important clues to the diagnosis. In addition to their generally globular shape, the tumors are truly encapsulated and an associated parent nerve is often apparent. The cut surface reveals a subcapsular zone of smooth, slightly dull, homogeneously light tan and firm tissue. Entrapped intratumoral axons are few, often subcapsular, and seen only on special stains. Gross features include globular enlargement of a fascicle (A), yellow coloration of the cut surface due to lipid accumulation (B), hemorrhage within the substance of the tumor (C), and cystic transformation (D). Schematic showing origin of a tumor within a fascicle, its expansion, and the incorporation of occasional axons within the substance of the lesion. The spindle cells contribute to Verocay body (A) or to whorl formation (B), as well as loose-textured Antoni B tissue (C) that is prone to microcystic change (D), hyalinized and thrombosed vessels (E), and perivascular iron deposition (F). Degenerative nuclear atypia, although common in large, longstanding tumors, is of no clinical significance (A). At the gross level, confusion may also arise with hypervascular tumors, such as hemangioma and paraganglioma. A helpful diagnostic gross feature is the presence of homogeneously light tan, firm tissue. Ultrastructurally the tumor shows processes coated with continuous basal lamina (arrows), as well as the formation of long spacing collagen or Luse bodies (asterisk). This encapsulated, sizable lesion shows focal hemorrhage related in part to prior biopsy. Conventional schwannomas are benign tumors that can be cured by complete resection. When incompletely excised, they occasionally recur and become clinically symptomatic. Effective surgical treatment of schwannomas needs to include within the resection specimen, the entering and exiting nerve fascicle, which can often be separated from the parent nerve. There is no compelling evidence that schwannomas undergo accelerated growth during pregnancy, and as such, surgery can typically be delayed until after delivery. The exceptionally rare occurrence of malignant transformation in conventional schwannoma is discussed later in the chapter. Although there are subtle cytologic differences between cellular schwannomas and smooth muscle tumors, a few simple observations aid in this distinction. Smooth muscle tumors are typically unencapsulated, whereas cellular schwannomas feature a capsule and are invariably strongly and diffusely reactive for S-100 protein, but immunonegative for muscle markers. Antoni A tissue predominates in cellular schwannoma by definition, the cells being arranged in fascicles or whorls; hyperchromasia and nuclear pleomorphism are generally mild. Hyalinized blood vessels and subcapsular lymphoid deposits are also common features. Features include encapsulation and composition of Antoni A tissue (A), degenerative vascular changes (B), increased cellularity and mitotic figures (arrows) compared to conventional schwannoma (C), and subcapsular chronic inflammation (D). Cellular schwannomas are benign but capable of recurrence when incompletely excised. This is particularly true of proximal tumors involving the spinal nerves extending into the spinal canal, sacral tumors, and intracranial examples. Plexiform Schwannoma Incidence, Demographics, Clinical Manifestations, and Localization. Roughly 5% of all schwannomas are multinodular or plexiform and of either conventional or cellular subtype. In a large study, head and neck (23%) and skin (15%) were found to be most often affected. Only occasionally do they show the entwining "worms" appearance of plexiform neurofibroma. As a rule, lymphoid aggregates and clusters of lipid-laden histiocytes are absent. In small tumors, entrapped axons are few or absent on neurofilament protein stain but are more readily seen in larger examples. Although such tumors may recur locally, both their immunohistochemistry and lack of metastatic potential argue in favor of their status as cellular schwannoma subtypes. Plexiform neurofibromas21 are far less cellular, contain more mucinous matrix, and show fewer S-100 protein staining cells. Like most plexiform schwannomas, neurotropic melanomas occur in the skin and subcutaneous tissue. Given profound differences in prognosis, it is of utmost importance to distinguish the two. Neurotropic melanoma favors the head and neck and is often associated with an overlying lentigo-type in situ melanoma. Both tumors involve nerves, but plexiform schwannomas form a mass pushing the distorted, remnant parent nerve to one side, such that residual normal nerve is often difficult to identify. Neurotropic melanomas primarily occupy the epineurium and encase as well as invade the perineurium to form distinctive, concentric rings surrounding intact endoneurium. Additionally, neurotropic melanomas have nuclei that are larger, more irregular in shape, and more often markedly hyperchromatic. Lastly, neurotropic melanoma is commonly associated with prominent desmoplasia, whereas plexiform schwannoma is not. As mentioned, however, a small proportion of incompletely excised tumors do recur. Tumors of Peripheral Nerve Epithelioid Schwannoma Both neurofibromas and schwannomas may show epithelioid features. Most are subcutaneous and behave in a benign fashion, even when atypical features. Malignant Transformation of Conventional, Cellular, and Plexiform Schwannoma Of conventional, cellular, and plexiform schwannomas, only the first two, on very rare occasion, have been found to undergo malignant transformation. The malignant epithelioid cells possess abundant eosinophilic cytoplasm, vesicular nuclei, and prominent nucleoli. Also of note are rare schwannomas containing clusters of atypical epithelioid cells with nuclear hyperchromasia and small nucleoli. Sarcomatous differentiation of the Schwann cell component of a schwannoma is extremely rare and may take the form of rhabdomyosarcoma. The term melanotic schwannoma is likely a misnomer, and most now consider this tumor to be a unique diagnostic entity, rather than merely a histologic variant of schwannoma. This very uncommon, clinicopathologically and genetically unique tumor37 has a predilection for posterior spinal nerves and their ganglia. Its peak incidence is in the fourth decade, and in contrast to schwannomas a subset of these tumors metastasize.
Order 40 mg valsartan mastercard
Moreover blood pressure 80 over 60 order cheap valsartan line, they occasionally may simulate the radiographic and pathologic appearances of selected mesothelial neoplasms or metastases of malignant tumors in the pleural space. Asbestos fibers are absent in the plaques themselves, and, if present, are seen only in the subjacent pulmonary parenchyma. Unilateral plaques may also be seen in patients with asbestos exposure, but these plaques may also develop as a consequence of chronic pleural irritation of any type. They are commonly associated with infections such as tuberculosis or empyema, chronic or recurrent pleural hemorrhage, and chest wall trauma. Those lesions associated with occupational asbestos exposure show an average latency of 20 years or more from the time of initial dust inhalation. It represents a pseudotumoral fibrohyaline pleural plaque in a patient with occupational-level asbestos exposure. These changes likely reflect the proposed mechanism of formation of the lesion: namely, that of recurrent and organizing pleuritis. Their possible relationship to bronchogenic carcinoma or mesothelioma has been examined, as reviewed elsewhere. The medium-power image demonstrates a vaguely lamellated appearance; capillaries are relatively numerous and oriented vertically to the pleural surface. It may be associated with connective tissue disorders, such as lupus erythematosus or rheumatoid arthritis, as well as chronic infections and asbestos exposure. Inflammatory myofibroblastic tumor: cytogenetic evidence supporting clonal origin. Pulmonary hamartoma: a clinical study of 77 cases in a 21 year period and review of the literature. An intrapulmonary chondromatous hamartoma penetrating the visceral pleura: report of a case. Placental transmogrification of the lung is a histologic pattern frequently associated with pulmonary fibrochondromatous hamartoma. Cytologic features of pulmonary hamartoma: report of a case diagnosed by fine needle aspiration cytology. Fine-needle aspiration of pulmonary hamartoma: a common source of false-positive diagnoses in the College of American Pathologists Interlaboratory Comparison Program in Nongynecologic Cytology. Recombinations of chromosomal bands 6p21 and 14q24 characterize pulmonary hamartomas. Clonal rearrangement of chromosome band 6p21 in the mesenchymal component of pulmonary chondroid hamartoma. Endometrial stromal sarcomas with unusual histologic features: a report of 24 primary and metastatic tumors emphasizing fibroblastic and smooth muscle differentiation. Metaplastic breast carcinoma metastatic to lung mimicking a primary chondroid lesion: report of a case with cytohistologic correlation. Salivary gland-type tumors with myoepithelial differentiation arising in pulmonary hamartoma: report of 2 cases of a hitherto-unrecognized association. Inflammatory pseudotumors of the lung: progression from organizing pneumonia to fibrous histiocytoma or to plasma cell granuloma in 32 cases. Calcifying fibrous pseudotumor of the pleura: a report of three cases of a newly described entity involving the pleura. Calcifying fibrous pseudotumor arising within an inflammatory pseudotumor: evidence of progression from one lesion to the other Inflammatory pseudotumor (plasma cell granuloma) of lung, liver, and other organs. Plasma cell granuloma involving the tracheobronchial angle in a child: a case report. Plasma cell granuloma of the lung in childhood: atypical radiologic findings and association with hypertrophic osteoarthropathy. Inflammatory pseudotumor of the lung: clinicopathological analysis in seven adult patients. Distinctive pulmonary histopathology with increased IgG4-positive plasma cells in patients with autoimmune pancreatitis: report of 6 and 12 cases with similar histopathology. Inflammatory myofibroblastic tumor versus IgG4related sclerosing disease and inflammatory pseudotumor: a comparative clinicopathologic study. Presence of immunoglobulin heavy-chain rearrangement in so-called "plasma cell granuloma" of the lung. Primary pulmonary lymphoma-clinical review from a single institution in Singapore. Epithelial plasma cell granuloma-like tumors of the lungs: a hithertounrecognized tumor. Inflammatory sarcomatoid carcinoma of the lung: report of three cases and comparison with inflammatory pseudotumors in adult patients. Expression of thyroid transcription factor-1 and other markers in sclerosing hemangioma of the lung. Corticosteroids in the management of unresected plasma cell granuloma (inflammatory pseudotumor) of the lung. Treatment of plasma cell granuloma of the lung with radiation therapy: a report of two cases and a review of the literature. Pseudotumor resulting from atypical mycobacterial infection: a "histoid" variety of Mycobacterium avium-intracellulare complex infection. Spindle cell pseudotumors in the lungs due to Mycobacterium tuberculosis in a transplant patient. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): report of a case with widespread nodal and extranodal dissemination. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): three unusual manifestations. Pulmonary extramedullary hematopoiesis in patients with myelofibrosis undergoing allgeneic stem cell transplantation. Fine needle aspiration diagnosis of extramedullary hematopoiesis presenting as a right lung mass. A 62 year-old woman with bilateral pleural effusions and pulmonary infiltrates caused by extramedullary hematopoiesis. A study on neoplastic cells in sputum as a contribution to the diagnosis of primary lung cancer. Cytopathologic diagnosis of pulmonary neoplasms in sputum and bronchoscopic specimens. Experience with the cytologic detection, localization, and treatment of radiographically undemonstrable bronchial carcinoma. Diagnostic fiberoptic bronchoscopy: techniques and results of biopsy in 600 patients. Diagnosis and typing of lung carcinomas by cytopathologic methods: a review of 108 cases. Bronchiolar proliferation and metaplasia associated with bronchiectasis, pulmonary infarct, and anthracosis. Bronchiolar proliferation and metaplasia associated with thromboembolism: a pathological and experimental study. Cytological evaluation of the sputum in patients with bronchiectasis and the possibility of erroneous diagnosis of carcinoma. Pulmonary tumorlets in cases of "tuberculoma" of the lung with malignant cells in brush biopsy. False-positive interpretations in respiratory cytopathology: exemplary cases and literature review. Cytologic changes of the respiratory tract in young adults as a consequence of high levels of air pollution exposure. Cytology of hyperplastic and neoplastic lesions of terminal bronchioles and alveoli. Relationship of interstitial pneumonia and honey-combing and typical epithelial proliferation to cancer of the lung. Alveolar metaplasia: its relationship to pulmonary fibrosis in industry and development of lung cancer.
