

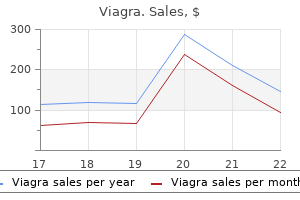
Cheap 100mg viagra otc
However constipation causes erectile dysfunction cheap viagra 100 mg, quantitative viral loads that could be used as trigger points for intervention remain undefined at a global level because of lack of assay standardization and few natural history studies with sufficiently large patient populations. Along with the absolute viral load, the kinetics of viral load increase may play an important role in defining risk. However, it has poor specificity, resulting in good negative (greater than 90%) but poor positive (as low as 28% and not greater than 65%) predictive values in these populations. Monitoring in plasma may have better specificity than whole-blood monitoring when used in this setting. In contrast, when rituximab was used, viral load measured in cellular blood components often fell dramatically and remained low even when disease progressed and relapsed (144, 145). There are some preliminary data to suggest that plasma may be preferable to whole blood when used to assess treatment response and predict relapse. The definitive diagnosis of this condition requires genetic studies to detect specific associated hereditary mutations. Prolonged inhibitory effect of 9-(1,3-dihydroxy-2-propoxymethyl)guanine against replication of Epstein-Barr virus. Effect of acyclovir on infectious mononucleosis: a double-blind, placebocontrolled study. Acyclovir and prednisolone treatment of acute infectious mononucleosis: a multicenter, double-blind, placebocontrolled study. A cohort study among university students: identification of risk factors for EpsteinBarr virus seroconversion and infectious mononucleosis. Clinical features and treatment strategies of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Pathogenesis of chronic active EpsteinBarr virus infection: is this an infectious disease, lymphoproliferative disorder, or immunodeficiency? Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 89 September 2008. Kimura H, Morishima T, Kanegane H, Ohga S, Hoshino Y, Maeda A, Imai S, Okano M, Morio T, Yokota S, Tsuchiya S, Yachie A, Imashuku S, Kawa K, Wakiguchi H. Kimura H, Ito Y, Kawabe S, Gotoh K, Takahashi Y, Kojima S, Naoe T, Esaki S, Kikuta A, Sawada A, Kawa K, Ohshima K, Nakamura S. Capacity of Epstein-Barr virus to infect monocytes and inhibit their development into dendritic cells is affected by the cell type supporting virus replication. Detection and quantification of latently infected B lymphocytes in Epstein-Barr virus-seropositive, healthy individuals by polymerase chain reaction. Behavioral, virologic, and immunologic factors associated with acquisition and severity of primary EpsteinBarr virus infection in university students. Detection of multiple "Ebnotypes" in individual Epstein-Barr virus carriers following lymphocyte transformation by virus derived from peripheral blood and oropharynx. Long-term shedding of infectious Epstein-Barr virus after infectious mononucleosis. X-linked lymphoproliferative disease: clinical, diagnostic and molecular perspective. Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Epstein-Barr viral load as a tool to diagnose and monitor post-transplant lymphoproliferative disease. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplantation. Epstein-Barr virus-associated smooth muscle tumors are distinctive mesenchymal tumors reflecting multiple infection events: a clinicopathologic and molecular analysis of 29 tumors from 19 patients. Jonigk D, Laenger F, Maegel L, Izykowski N, Rische J, Tiede C, Klein C, Maecker-Kolhoff B, Kreipe H, Hussein K. Molecular and clinicopathological analysis of Epstein-Barr virus-associated posttransplant smooth muscle tumors. Oral hairy leukoplakia: clinicopathologic features, pathogenesis, diagnosis, and clinical significance. Chronic high Epstein-Barr viral load state and risk for late-onset posttransplant lymphoproliferative disease/ lymphoma in children. Chronic high Epstein-Barr viral load carriage in pediatric small bowel transplant recipients. Chronic high Epstein-Barr viral load carriage in pediatric liver transplant recipients. Bekker V, Scherpbier H, Beld M, Piriou E, van Breda A, Lange J, van Leth F, Jurriaans S, Alders S, Wertheimvan Dillen P, van Baarle D, Kuijpers T. Antibodies to Epstein-Barr virus thymidine kinase: a characteristic marker for the serological detection of nasopharyngeal carcinoma. Epstein Barr virus-associated tumours: an update for the attention of the working pathologist. Kimura H, Miyake K, Yamauchi Y, Nishiyama K, Iwata S, Iwatsuki K, Gotoh K, Kojima S, Ito Y, Nishiyama Y. Reduction of immunosuppression as initial therapy for posttransplantation lymphoproliferative disorder. A phase I trial of EpsteinBarr virus gp350 vaccine for children with chronic kidney disease awaiting transplantation. American Society of Transplantation recommendations for screening, monitoring and reporting of infectious complications in immunosuppression trials in recipients of organ transplantation. A prospective clinical study of Epstein-Barr virus and host interactions during acute infectious mononucleosis. Management of T-cell and natural-killer-cell neoplasms in Asia: consensus statement from the Asian Oncology Summit 2009. Ishii H, Ogino T, Berger C, Kochli-Schmitz N, Nagato T, Takahara M, Nadal D, Harabuchi Y. Comparison of various blood compartments and reporting units for the detection and quantification of Epstein-Barr virus in peripheral blood. Comparison of six different specimen types for Epstein-Barr viral load quantification in peripheral blood of pediatric patients after heart transplantation or after allogeneic hematopoietic stem cell transplantation. Cytomegalovirus and Epstein-Barr virus subtypes-the search for clinical significance. Evaluation of 12 commercial tests for detection of Epstein-Barr virus-specific and heterophile antibodies. Routine Epstein-Barr virus diagnostics from the laboratory perspective: still challenging after 35 years. Evaluation of a multianalyte profiling assay and an enzyme-linked immunosorbent assay for serological examination of Epstein-Barr virus-specific antibody responses in diagnosis of nasopharyngeal carcinoma. Rapid detection of antibodies in sera using multiplexed self-assembling bead arrays. Evidence-based approach for interpretation of EpsteinBarr virus serological patterns. Evaluation of four commercially available Epstein-Barr virus enzyme immunoassays with an immunofluorescence assay as the reference method. Evaluation of three commercial enzyme-linked immunosorbent assays and two latex agglutination assays for diagnosis of primary Epstein-Barr virus infection. No correlation in Epstein-Barr virus reactivation between serological parameters and viral load. Evaluation of enzymelinked immunosorbent assays with two synthetic peptides of Epstein-Barr virus for diagnosis of infectious mononucleosis. Simplicity through complexity: immunoblot with recombinant antigens as the new gold standard in Epstein-Barr virus serology. Prevention of Epstein-Barr viruslymphoproliferative disease by molecular monitoring and preemptive rituximab in high-risk patients after allogeneic stem cell transplantation. Epstein-Barr virus load in whole blood is associated with immunosuppression, but not with post-transplant lymphoproliferative disease in stable adult heart transplant patients. Serial measurement of Epstein-Barr viral load in peripheral blood in pediatric liver transplant recipients during treatment for posttransplant lymphoproliferative disease. Epstein-Barr viral load in whole blood of adults with posttransplant lymphoproliferative disorder after solid organ transplantation does not correlate with clinical course.
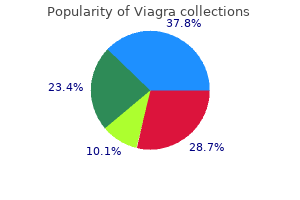
Buy viagra american express
Many include an internal control that targets a human housekeeping gene erectile dysfunction protocol free ebook purchase viagra from india, such as -globin. Some type-specific differences in amplification efficiency have been noted for several tests. Additionally, the selection of a genotyping test is dependent on the source material. Broad-spectrum genotyping assays are useful primarily for research purposes, including postvaccination genotype distribution surveillance. Laboratorians should encourage following the current testing guidelines with respect to patient age, cotesting, reflex testing, and testing frequency. There will be an emerging literature on the role of epigenetics as potential diagnostic tools that merit further observation. Assays must be robust and highly reliable and should display inter- and intralaboratory reproducibility. Additionally, the data confirmed that screening intervals can be extended to at least 5 years. Genital human papillomavirus infection in men: incidence and risk factors in a cohort of university students. A review of the evidence comparing the human papillomavirus vaccine versus condoms in the prevention of human papillomavirus infections. Modeling the sexual transmissibility of human papillomavirus infection using stochastic computer simulation and empirical data from a cohort study of young women in Montreal, Canada. Genital human papillomavirus infection: incidence and risk factors in a cohort of female university students. Risk of female human papillomavirus acquisition associated with first male sex partner. Patterns of persistent genital human papillomavirus infection among women worldwide: a literature review and meta-analysis. Longterm absolute risk of cervical intraepithelial neoplasia grade 3 or worse following human papillomavirus infection: role of persistence. Evaluation of any or type-specific persistence of high-risk recommendation from the U. Gene expression of genital human papillomaviruses and considerations on potential antiviral approaches. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. Vinokurova S, Wentzensen N, Kraus I, Klaes R, Driesch C, Melsheimer P, Kisseljov F, Durst M, Schneider A, von Knebel Doeberitz M. Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Integration of high-risk human papillomavirus: a key event in cervical carcinogenesis? Acquisition of high-level chromosomal instability is associated with integration of human papillomavirus type 16 in cervical keratinocytes. The natural history of human papillomavirus type 16 capsid antibodies among a cohort of university women. Serum antibody response following genital 9 human papillomavirus infection in young men. A competitive serological assay shows naturally acquired immunity to human papillomavirus infections in the Guanacaste Natural History Study. Seroprevalence of 8 oncogenic human papillomaviruses and acquired immunity against re-infection. Natural history of anal human papillomavirus infection in heterosexual women and risks associated with persistence. Impact of human immunodeficiency virus on the natural history of human papillomavirus genital infection in South African men and women. Auvert B, Sobngwi-Tambekou J, Cutler E, Nieuwoudt M, Lissouba P, Puren A, Taljaard D. Effect of male circumcision on the prevalence of high-risk human papillomavirus in young men: results of a randomized con- 1797 32. The epidemiology and natural history of anal human papillomavirus infection in men who have sex with men. Cervical intraepithelial neoplasia outcomes after treatment: long-term follow-up from the British Columbia Cohort Study. Post treatment human papillomavirus testing for recurrent cervical intraepithelial neoplasia: a systematic review. Human papillomavirus related head and neck cancer survival: a systematic review and metaanalysis. Self-collected samples for testing of oncogenic human papillomavirus: a systematic review. Accuracy of human papillomavirus testing on self-collected versus clinician-collected samples: a meta-analysis. Looking ahead: a case for human papillomavirus testing of self-sampled vaginal specimens as a cervical cancer screening strategy. Cytologic detection of cervical abnormalities using liquid-based compared with conventional cytology: a randomized controlled trial. Comparison of liquid-based cytology with conventional cytology for detection of cervical cancer precursors: a randomized controlled trial. Roelens J, Reuschenbach M, von Knebel Doeberitz M, Wentzensen N, Bergeron C, Arbyn M. Comparison of human papillomavirus testing strategies for triage of women referred with lowgrade cytological abnormalities. Commercially available assays for multiplex detection of alpha human papillomaviruses. Overview of human papillomavirus-based and other novel options for cervical cancer screening in developed and developing countries. Restricted cross-reactivity of hybrid capture 2 with nononcogenic human papillomavirus types. Ratnam S, Coutlee F, Fontaine D, Bentley J, Escott N, Ghatage P, Gadag V, Holloway G, Bartellas E, Kum N, Giede C, Lear A. Immunogenicity testing in human papillomavirus virus-like-particle vaccine trials. In situ hybridization detection of single-copy human papillomavirus on isolated cells, using a catalyzed signal amplification system: GenPoint. Detection and genotyping of human papillomavirus by five assays according to cytologic results. Comparison of predictors for high-grade cervical intraepithelial neoplasia in women with abnormal smears. Comparison of seven tests for highgrade cervical intraepithelial neoplasia in women with abnormal smears: the Predictors 2 study. Cuzick J, Cadman L, Mesher D, Austin J, AshdownerBarr L, Ho L, Terry G, Liddle S, Wright C, Lyons D, Szarewski A. Comparing the performance of six human papillomavirus tests in a screening population. Type-specific detection of 30 oncogenic human papillomaviruses by genotyping both E6 and L1 genes. Alaghehbandan R, Fontaine D, Bentley J, Escott N, Ghatage P, Lear A, Coutlee F, Ratnam S. Benevolo M, Vocaturo A, Caraceni D, French D, Rosini S, Zappacosta R, Terrenato I, Ciccocioppo L, Frega A, Giorgi Rossi P. Clinical validation of AdvanSure GenoBlot assay as primary screening and test of cure for human papillomavirus infection. Chranioti A, Spathis A, Aga E, Meristoudis C, Pappas A, Panayiotides I, Karakitsos P. Bryant D, Rai N, Rowlands G, Hibbitts S, Jones J, Tristram A, Fiander A, Powell N. Comparison of PapType to Digene Hybrid Capture 2, Roche Linear Array, and Amplicor for detection of high-risk human papillomavirus genotypes in women with previous abnormal pap smears. Individual detection of 14 high risk human papilloma virus genotypes by the PapType test for the prediction of high grade cervical lesions.
| Comparative prices of Viagra | ||
| # | Retailer | Average price |
| 1 | O'Reilly Automotive | 126 |
| 2 | McDonald's | 182 |
| 3 | Winn-Dixie Stores | 963 |
| 4 | Gap | 488 |
| 5 | Michaels Stores | 858 |
| 6 | Family Dollar | 956 |
| 7 | Costco | 776 |
| 8 | ShopRite | 511 |
| 9 | Kroger | 710 |
| 10 | J.C. Penney | 243 |
Buy viagra in india
Most current testing is still performed on citrated blood within a few hours of sampling erectile dysfunction cancer order viagra american express. Global tests of platelet function are often initially used as screening tests during the laboratory investigation of individuals 711 Postgraduate Haematology with suspected haemostatic defects. Since global tests of platelet function do not enable specific diagnosis of platelet disorders, they are normally performed as the first part of a two-step strategy that requires further testing with more specialized assays of platelet function to confirm or refute any clinical diagnosis. The most commonly proposed rationale for testing global platelet function as a first-line investigation is exclusion of a platelet function disorder so that further specialized testing can be avoided. The bleeding time was the first in vivo test of platelet function and is performed by timing the arrest of bleeding from standard-sized cuts made in the skin of the forearm. Most clinicians consider the test to be poorly reproducible, invasive, insensitive (particularly to mild platelet defects) and timeconsuming. A number of in vitro tests that attempt to accurately simulate platelet function have been developed. Automated cell counting of whole blood in the modern full blood count investigation is an essential screening test in patients with abnormal bleeding. Normal results will therefore rapidly eliminate thrombocytopenia and anaemia as potential causes of bleeding and ensure that any platelet function tests that are being performed will not be affected by low platelet counts. Depending on the results of clinical and laboratory screening of patients, a series of diagnostic platelet function tests are usually performed. Conventional Born aggregometers monitor the changes in light transmission that occur in a suspension of platelets in plasma or a physiological buffer that are stimulated with different concentrations of various agonists. These parameters, coupled with the pattern of primary and secondary aggregation responses obtained with different dosages of different agonists, enable the experienced operator to diagnose specific signalling defects. Measurement of the storage and release of the dense granular nucleotides can also be performed by a variety of alternative methods for confirming either storage and/or release defects, although recent surveys suggest that measurement of secretion is underused. Whole-blood impedancebased aggregometers are now also common and measure the changes in electrical resistance between two electrodes caused by platelet adhesion and aggregation after addition of agonists, but are heavily influenced by platelet count. Flow cytometry provides an exquisite, sensitive and powerful tool for studying and diagnosing various platelet defects. Flow cytometric analysis of platelets is performed in fresh whole blood or in platelet-rich plasma, and the technique can be used with very small fluid volumes, even in thrombocytopenia. This technique is used to determine the copy density of platelet membrane glycoproteins and receptors, and is therefore useful for confirming the absence of various glycoproteins or receptors in disease. Platelet function testing can also be performed and the ability of platelets to degranulate, express activation markers. A major limitation of the above tests is that they are performed at low shear conditions and therefore do not mimic accurately many of the important physiological processes of platelet adhesion, activation and aggregation that occur at higher shear rates in vivo. However, a variety of screening and diagnostic tests coupled with an accurate bleeding history will always be required to confirm a platelet-based bleeding disorder, simply because of the heterogeneity in the range of disorders encountered coupled with the cross-talk between receptors. This could potentially allow patients to be risk stratified and their treatment adjusted accordingly to potentially improve their clinical outcomes. The test has also been adapted to measure aspirin and P2Y12 blockade with two other cartridge formulations. Emerging evidence suggests that the true incidence of so-called aspirin resistance is probably very low (when non-compliance is accounted for) and that many studies are measuring platelet hyper-reactivity and not blockade of cyclo-oxygenase type 1. Although recent meta-analyses suggest that this could nevertheless be clinically informative, it is still unclear how to reliably identify these patients, with which test and cut-off (as different tests give different results) and then actually how to manage these patients. Certainly there is a spectrum of platelet responsiveness within the normal population and those individuals with a stable or even an acquired hyper-reactive phenotype could theoretically be at higher risk of thrombosis. Clopidogrel resistance is a different phenomenon as this is a prodrug that requires metabolism by liver cytochrome P450. This results in the generation of an active metabolite that irreversibly inhibits the P2Y12 receptor. Differences in the efficiency of metabolism between individuals (related to their P450 genotype) taking standard clopidogrel dosing results in a spectrum of responsiveness and recent meta-analysis suggests that poor responders are indeed prone to increased risk of thrombosis. One of the problems facing the investigator is not only which test to use, but also the definition of a suitable cut-off defining a non-response and how to effectively manage these individuals. Although there are emerging clinical cardiology guidelines, as yet they do not include routine monitoring of clopidogrel responsiveness. It is clear that large well-designed prospective trials are required in this area, where patients with hyper-reactive platelets and/or a poor response to aspirin/clopidogrel are randomized to different treatments based on a platelet function test result. Only with this type of data will personalized platelet function testing perhaps become a reality in the future. Inherited platelet disorders can be classified into distinct groups, which include defects in adhesion, receptor signalling, secretion, cytoskeleton, procoagulant activity and production. This is due to the variable clinical expression of the bleeding symptoms and the relative redundancy of known platelet receptor and signalling pathways. This is in addition to a homozygous frameshift mutation in multiple Chilean patients, which is likely to be due to a founder effect. Only three functionally disrupting mutations have been reported in the Gq-coupled thromboxane receptor, of which all patients are heterozygous for the mutation. Since then, cloning 713 Postgraduate Haematology techniques have gone on to scrutinize the domains of genes which behold their function. Alternatively a more gene-specific targeted platform may be the method of choice to reduce the time to diagnosis, by allowing the capture and sequencing of a panel of genes of relevance to bleeding and thrombotic disorders. Insights into the underlying mechanisms of platelet function through genetic investigation in patients will, in turn, lead to more effective treatments for bleeding and cardiovascular disease in the future. New medicines targeted to the very early stages of platelet activation or the amplification phase (as proven with aspirin and clopidogrel) may be more effective in preventing thrombosis without significantly disturbing normal haemostasis. What is the role of genetic testing in the investigation of patients with suspected platelet function disorders Conclusions and future developments Given that platelets were considered to be just pieces of circulating dust about 150 years ago, they are now one of the most important cells in pathology. Now that the megakaryocytic/platelet genome and proteomes are being defined, many new platelet proteins have been discovered, although many of these appear to have minor or negligible roles in regulating activation. The challenge is to identify which of these are functionally important in the many roles of platelets. The ultimate goal of platelet research remains the development of improved antithrombotic agents that provide effective treatment without an increased risk of bleeding. Early writers were impressed by the helplessness of the physician in the face of haemophilic bleeding. Although long recognized, the pathophysiology and genetics of these disorders was not fully understood until the latter half of the twentieth century. Advances in protein chemistry and molecular biology now allow a comprehensive understanding of normal coagulation, the physiological defect in haemophilia and the underlying molecular genetics. In both cases, failure to form the intrinsic tenase complex results in failure to produce the thrombin burst characteristic of normal coagulation. As a result, a loose friable fibrin mesh is produced that is easily dislodged and which has increased susceptibility to fibrinolysis. The consequent failure to consolidate the primary haemostatic (platelet) plug results in the characteristic bleeding pattern of haemophilia, which is both delayed after trauma and much prolonged. Replacement by intravenous infusion of the deficient factor can normalize the haemostatic mechanism. Clinical features Haemophilia A and B affect approximately 1 in 10,000 and 1 in 50,000 live births, respectively, and are equally common in all ethnic groups. The vast majority of patients are male, but haemophilia can occur very rarely in females (see below). The main load- or strain-bearing joints (ankles, knees and elbows) are most affected, but any joint can be the site of bleeding. If untreated, this intracapsular bleeding causes severe swelling, pain, stiffness and inflammation, which gradually resolves over days or weeks. Combined with the fact that they are both encoded on the long arm of the X chromosome, they present almost indistinguishable sex-linked clinical syndromes and specific assays are required Postgraduate Haematology, Seventh Edition. No doubt, minor tissue damage consequent on everyday activities actually initiates bleeding. Bleeding into muscles and joints is the hallmark of haemophilia, although the pattern of bleeding described below is now often masked by the use of prophylaxis.
Viagra 100mg amex
Platelet transfusions Platelet transfusions are indicated for the prevention and treatment of haemorrhage in patients with thrombocytopenia or platelet function defects impotence natural food cheap 50 mg viagra otc. The largest group of patients, receiving up to 67% of all platelet concentrates, are haematology patients. The decision to transfuse platelets should be based on clinical assessment, taking into account clinical risk factors for bleeding and the extent and site of bleeding, together with the platelet count. The lack of evidence base supporting platelet transfusion practice has prompted increasing research in this area. Randomized controlled studies have attempted to tackle key questions around decision-making on prophylactic platelet transfusions in haemato-oncology patients. There is a lack of evidence for platelet thresholds, indicating the need for transfusion, in other clinical settings and, in particular, prior to procedures or surgery, with the use of empirical guidance to support practice. Platelet transfusion response and refractoriness the efficacy of platelet transfusions can be assessed by monitoring clinical response and by checking the platelet count increment. Immediate (10 to 60 minutes) and 24-hour post-transfusion platelet increments should be measured for evidence of effectiveness of therapy. Cryoprecipitate is indicated for fibrinogen replacement largely in the major haemorrhage setting (see below). Red cells are necessary for their oxygencarrying capacity and also because they contribute to improved haemostasis through a rheological effect leading to axial flow and therefore margination of platelets. Metabolic changes in stored blood include low pH, hypocalcaemia and hyperkalaemia. Although, theoretically, excess citrate in transfused blood could cause toxicity, its metabolism in the liver is usually rapid. In practice, the only situations when citrate toxicity is a real problem is with extremely rapid transfusion (one unit every 5 min), or in infants, especially if premature, having exchange transfusion with blood stored in citrate for longer than 5 days. Hypocalcaemia and hyperkalaemia are usually transient and rapidly corrected once the transfused blood is circulating. Acidosis is not generally significant, as citrate metabolism leads to an alkalosis. However, if a patient is severely shocked and under-transfused, acidosis may be a clinical problem. Cardiac irregularities, in particular ventricular fibrillation, may result from transfusion of large quantities of cold blood. The optimal functioning of coagulation factors and of platelets is also temperature dependent and effectiveness is reduced by hypothermia. Thus, the use of a blood warmer and keeping the patient warm are important measures in the management of patients with major haemorrhage. Approximately one-quarter of patients bleeding with severe trauma present with clotting abnormalities, indicating that the coagulopathy is not merely dilutional related to infusion of fluids or red cells. Indeed, coagulopathy associated with massive haemorrhage is likely to be multifactorial with contributory factors, including activation of fibrinolysis and consumptive coagulopathy, exacerbated by hypothermia and hypocalcaemia. The use of anticoagulant/antiplatelet drugs prior to surgery may further contribute to bleeding post surgery together with the heparin needed for bypass for cardiac surgery. The practical management of major haemorrhage in any setting needs a coordinated multidisciplinary approach. This should incorporate significant advances in techniques for resuscitation as well as surgical, radiological and endoscopy interventions to control bleeding. Tranexamic acid may also have benefits in major obstetric haemorrhage and acute upper gastrointestinal haemorrhage, with multicentre clinical trials ongoing in these areas. Baseline blood samples should be taken for full blood count, chemistry, coagulation screen, and group and screen. In patients with severe haemorrhage the initial use of group O red cells is indicated before the blood group is available; RhD negative units should be given to females of childbearing age. There may be significant delays in obtaining laboratory clotting tests to initiate therapy. Maternal IgG can cross the placenta, and thus IgG1 and IgG3 red cell alloantibodies can gain access to the fetus. If the fetal red cells contain the corresponding antigen, then binding of antibody to red cells will occur. The effects on the fetus/newborn infant may vary according to the characteristics of the maternal alloantibody. Furthermore, the Lewis antigens are not red cell antigens per se and are not fully developed at birth. Some antibodies (particularly anti-D, -K and -c) are associated with significant fetal and neonatal risks, such as anaemia requiring intrauterine or neonatal transfusion, jaundice or perinatal loss due to hydrops fetalis or kernicterus. There are many antibodies that are unlikely to significantly affect the fetus, but can cause neonatal anaemia and hyperbilirubinaemia), while others may cause problems for the screening and timely provision of appropriate blood for the mother or fetus/neonate. The levels of anti-D and antic are quantified using automated analysers, whereas the titre of all other antibodies is determined by doubling dilution. For anti-K, severe fetal anaemia can occur, even with low titres, so early referral to a fetal medicine specialist is advisable. Ultrasound monitoring may help in detecting features suggesting severe fetal anaemia including polyhydramnios, skin oedema and cardiomegaly. It is therefore essential to also determine the Hb and bilirubin level to ascertain the degree of anaemia and haemolysis at birth, as this helps guide management, either with phototherapy or transfusion (see also Chapter 50). While this may be due to passively acquired maternal antibodies causing continued haemolysis, it may also be due to suppression of erythropoiesis with extremely low reticulocytes in the affected infants and further top-up transfusions may be needed. Kleihauer) testing to check if further anti-D Ig needed, in addition to the standard dose, which should be given in the first instance after delivery. Reproduced with permission of the Royal College of Obstetricians and Gynaecologists. Following sensitising events, anti-D Ig should be administered as soon as possible and ideally within 72 hours of the event. If, exceptionally, this deadline has not been met, some protection may be offered if anti-D Ig is given up to 10 days after the sensitizing event. This can be with a single dose regimen at 28 weeks, or two dose regimen given at 28 and 34 weeks. It is important that the 28-week sample for blood group and antibody screen is taken prior to the first routine prophylactic anti-D Ig injection. Peak velocity of systolic blood flow in the middle cerebral artery in 111 fetuses at risk for anaemia due to maternal red cell alloimmunization. Solid curve indicates the median peak systolic velocity in the middle cerebral artery and the dotted curve indicates 1. Good communication between the antenatal teams and the transfusion laboratory is essential, with clear documentation of issue and administration of anti-D immunoglobulin. The principle of the Kleihauer test relies on the different properties of fetal haemoglobin (HbF) and adult haemoglobin (HbA), whereby HbF is more resistant than HbA to both acid elution and alkaline denaturation. When a blood film from the maternal sample is fixed and immersed in an acid buffer solution, HbA is denatured and eluted, leaving red cell ghosts. Fetal red cells containing HbF that is resistant to elution stand out after staining in a background of maternal ghost cells. Flow cytometry is used to quantify the population of RhDpositive fetal cells in the RhD-negative maternal sample, using a fluorochrome-conjugated IgG monoclonal anti-D to label the D-positive cells.
Discount 100 mg viagra otc
Data regarding the use of hydroxycarbamide (hydroxycarbamide hydroxyurea) in this setting are scant and busulfan should be avoided impotence vs impotence cheap 75 mg viagra amex. Patients usually present with leucocytosis, mainly caused by increased number of neutrophils and their precursors. Shading of Redaelli: orange, highly resistant; yellow, resistant; blue, moderately resistant; and green, sensitive. The bulk of transplant outcome data came from patients who underwent myeloablative conditioning, which usually consists of high-dose cyclophosphamide and either busulfan or total body irradiation. Transplant-related mortality, graft-versus-host disease and relapse-related mortality are significant problems. A number of emerging biomarkers with prognostic significance are currently under investigation, and early identification of patients who are at high risk of transformation should become possible. At the other end of the spectrum, an increasing number of patients achieve stable and deep molecular responses. Acknowledgement the authors would like to thank Dr Agnes Yong for her critical review of this chapter. Prognostication is enabled by classification schemes, which take into account the diverse clinical features, and is further supported by scoring systems so that therapy can be both symptomatic and risk directed. While some of these anaemias were apparently attributable to coexisting chronic diseases, others arose de novo in otherwise healthy individuals. Bone marrow analysis often revealed a spectrum of abnormalities, notably aberrant maturation of marrow precursors, a variable increase in marrow blasts and, in some patients, the presence of ring sideroblasts. However, this rate increases markedly with age, exceeding 30 per 100,000 for individuals over the age of 80 years. Exposure to ionizing radiation, benzene, solvents, pesticides and smoking are implicated; however, case-control studies are inconsistent. The risk increases with age and with prolonged exposure to low-dose chemotherapy, such as prescribed for controlling vasculitic and other autoimmune disorders. Most cases develop within 5 years of transplantation, with the major risk factors being the duration and amount of pretransplant chemotherapy, whether the patient received total body irradiation and the dose of infused stem cells. The clinical and morphological features of this subgroup are distinct, occur more frequently in elderly women, and are characterized by Table 25. Erythropoiesis is often reduced, with mild to moderate dysplasia, and myeloblasts comprise less than 5% of the myelogram. Note loss of material from long arm of chromosome 5 and characteristic hypolobated megakaryocytes. Their role alongside the effect of the bone marrow microenvironment and the host immunity in the emergence and progression of the disease are discussed in the following section. If so, then this would support the multistep theory of disease progression by which the dysplastic clone evolves over time through acquisition of additional mutations, allowing selection of more proliferative subclones. Thus almost all cells of the bone marrow myeloid lineage (red cell, granulocytic/ megakaryocytic precursors and megakaryocytes) are clonal. The initial driver mutation may also shape the future trajectory of clonal evolution through constraints on the repertoire of the cooperating genetic events (see also Chapter 18). Interference with this niche by targeting the molecular interactions between the stem cell and its microenvironment represents a novel therapeutic strategy. Within this 442 environment myeloid-derived suppressor cells accumulate and inhibit autologous haemopoiesis. The interception of the ligand with soluble receptor or interruption of the signalling cascades mediates reversal in ineffective haemopoiesis and is an area of active study. For those patients undergoing leukaemic transformation, the cytopenias arise due to maturation block of the malignant cells. Indeed, for the blasts to overcome this apoptotic tendency indicates that they have lost their G2/M checkpoint control that appears to be a necessary requirement for progression to leukaemia. The complex group consisted of 66 patients of whom 63 had chromosome 5 and/or 7 abnormalities in addition to other abnormalities (abnormalities of chromosome 5 n = 33, chromosome 7 n = 7 and both 23). However, 14% of patients had cytogenetic abnormalities of unknown prognostic significance. If a cytogenetic anomaly occurred in at least five patients it was a distinct subgroup. Complex karyotypes are also further subdivided into those with three unrelated cytogenetic abnormalities and those with more than three karyotypic abnormalities. Independent clones where two subclones occur in parallel form a subgroup provided neither clone is complex. In studies of patients with myeloid neoplasms, 44% have abnormal metaphase cytogenetics, 49% have a normal karyotype and 7% are uninformative. The search for a minimal common deleted region on the long arm of chromosome 5 has spanned three decades and relied on physical mapping methods. The search for a tumour-suppressor gene located within this region has involved many different approaches over recent years and resulted in numerous candidate genes. These cells were cultured in conditions promoting either erythroid or megakaryocytic differentiation. It can present as a total or partial monosomy and may be an isolated abnormality or part of a complex karyotype. Monosomy 7 confers a poor prognosis that ranges from 14 months as an isolated abnormality down to 7 months if the karyotype is complex and involves other abnormalities. The relatively better prognosis of patients with large deletions and their occurrence in children suggests these may be founding lesions, whereas the smaller partial deletions of 7q represent a secondary event due to genomic instability. Deletions of 20q generally carry a favourable prognosis and the pathogenetic gene(s), within the commonly deleted region 20q12, are unknown. The mutations are heterozygous missense mutations due to A to G transversion, leading to substitution of lysine at residue 700 by glutamic acid (K700E) or C to A or C to G substitution at position 662, where histidine is replaced by glutamine (H662Q). These are heterozygous, missense mutations specifically at P95H (proline at position 95 which may be replaced by histidine, leucine, arginine, alanine or threonine). The rectangles and broken lines represent exonic and intronic regions, respectively. Crosstalk between spliceosome mutations and specific epigenetic modifiers has also been suggested. Such CpG pairs are underrepresented in the human genome, but cluster together within so-called CpG islands that tend to be located in the proximity of gene promoter regions. In normal cells, these CpG islands are typically unmethylated, allowing genes to be transcribed. However, if CpG islands are methylated, then transcriptional Chapter 25 the myelodysplastic syndromes activity at the promoter is impeded and the gene is silenced. Thus, aberrant promoter methylation leads to inactivation of the gene, thereby providing a mechanism where genes can be functionally inactive. Biochemical alterations to the tails of the histone molecules influence the degree of compaction of the nucleosomes and hence the level of transcriptional activity of nearby genes. Moreover, there is a close and cooperative interplay between these two epigenetic control mechanisms that together can render a gene permanently silenced. The significance of this is that combination epigenetic therapies that comprise a hypomethylating agent with a histone deacetylase inhibitor may be more effective than single agents in re-expressing silenced tumoursuppressor genes. The mutations are often nucleotide substitutions resulting in missense, frame shift, stop codons, in-frame deletions or amino acid substitutions in extremely conserved residues. Post-translational modifications of histones play an important part in epigenetic regulation of gene expression.
Syndromes
- Name of the product (ingredients and strengths if known)
- Difficulty understanding speech
- Is it worse after physical exertion?
- Helicobacter pylori infection of the stomach
- A starch item (such as potatoes)
- Thyroid biopsy
- Feeding difficulty (food may go into the windpipe instead of the stomach)
Best purchase for viagra
Hypogammaglobulinaemia is frequent (30% of patients) and tends to worsen over the course of the disease erectile dysfunction treatment medicine discount viagra line. Note the dual population of small lymphocytes and larger nucleolated prolymphocytes. In around 30% of cases, cytogenetic evolution occurs over the course of the disease. Bone marrow aspirate and biopsy are not necessary for diagnosis, but can be useful in cases where diagnosis is complex, and to provide invaluable information about the origin of cytopenias. Differential diagnosis Pathology features In typical cases, the histology of lymph nodes shows effacement of the architecture by small lymphocytes with a pseudofollicular pattern of pale areas that correspond to proliferation centre. Proliferation centre contain numerous T cells, a fine network of dendritic cells and B cells with an increased expression of proliferation-associated markers and antiapoptotic molecules compared to the non-proliferation component. The bone marrow displays a variable degree of infiltration by the disease and, in contrast to follicular lymphoma, there is no paratrabecular infiltration. Four infiltration patterns have been described: nodular, interstitial, mixed (nodular+interstitial) and diffuse. While in early clinical phase nodular and interstitial patterns predominate, in advanced phase a diffuse infiltration is the norm. The diagnosis requires the exclusion of other B-cell disorders with plasmatic differentiation. In particularly difficult cases, biopsy of involved lymph nodes and bone marrow, as well as genetic and molecular studies, can be of help (Table 27. In affected individuals, the absolute number of B cells is increased, but, as shown by / staining, they are polyclonal. These persons should not receive therapy, but be closely followed to detect a potential disease transformation. There are cases in which there is more than one B-cell clone, and even others in which a mixture of monoclonal B cells and monoclonal T cells can be detected. The diagnosis needs to be substantiated by the biopsy of a lymph node or another lymphoid tissue. Outcome predictors need to fulfill a number of requisites to be considered as such. For example: standardization, inter- and intrareproducibility, independent prognostic value from other predictors of the same outcome and direct consequences in the management of a substantial proportion of patients. Importantly, outcome predictors can complement, but not replace, clinical expertise and sound clinical judgment. This is due at least in part to better therapies, particularly in younger patients. The individual prognosis, however, is heterogeneous and ranges from a few months to a normal lifespan. Although somewhat overlapping, it is useful to distinguish, under the umbrella of outcome predictors, those parameters that predict disease progression, and hence need of therapy (prognostic factors), and those informing about the probability that an individual patient will respond to a given therapy (predictive factors) (Table 27. Whether this is because of the gender itself or the characteristics of the disease in women is unclear. Particularly important is an impaired renal function (creatinine clearance <70 mL/min), which is associated with poor tolerance to chemoimmunotherapy and higher toxicity. Clinical stages Although developed more than 30 years ago, the clinical stages independently formulated by Rai and Binet continue to be the backbone of estimating prognosis and indicating therapy. Rai and Binet clinical staging systems give reliable information on survival probability. Importantly, assigning a clinical stage to a given patient only requires a physical examination and a complete blood cell count; such simplicity is a tremendous advantage, enabling them to be applied globally. The most common abnormalities are del(13q), trisomy 12, del(11q), del(6q) and del(17p). This model implied that patients are classified according to the worst cytogenetic abnormality, as shown in Table 27. It should be noted that deletions of 13q14, present in up to 50% of cases, confer a good prognosis, provided they are found as the only change. Early reports suggested 30% as a cut-off, but more recent studies found lower figures (7%, 20%) to be more clinically relevant. This is particularly evident in patients with stage A disease, in whom early assessment of the need for future therapy may be desirable. Serum markers: -2 microglobulin (2M) has been extensively studied and validated from the prognostic standpoint. In many, but not all, studies it has been found to predict poor response to therapy and short survival. Prognostic systems and scores Prognosis is related to many factors, and thus a single parameter rarely predicts outcome. Because of this, prognostic assessment is based on the combination of different parameters in the form of stages, prognostic groups or scores. The best, and oldest, examples are Binet and Rai staging systems in which lymphadenopathy, organomegalies, anaemia and thrombocytopenia are combined to identify different risk groups (Table 27. Other systems to evaluate prognosis in either the whole patient population or selected groups have been proposed. Among elderly patients (>70 years old), the Barcelona group has shown that advanced stage (Binet B or C), increased serum 2M microglobulin (>2. Likewise, reticulocytosis may not be striking in the context of a bone marrow heavily infiltrated by leukaemic cells or in patients receiving treatment. Nevertheless, thrombocytopenia can be considered as immune mediated when platelet counts suddenly decrease in the absence of splenomegaly, infection or chemotherapy, and with abundant megakaryocytes in the bone marrow. Therefore, isolated thrombocytopenia, particularly if extremely low, is more likely to be immune in origin. In those areas in which this is a prevalent problem, Helicobacter pylori infection should be excluded. In the presence of anaemia, the reticulocyte percentage can be misleadingly normal; therefore, the reticulocyte percentage corrected according to the haematocrit and the absolute reticulocyte count are more informative. In the presence of a bone marrow heavily infiltrated by lymphocytes, the identification of red cell precursors can be difficult. In this setting, antiglycophorin immunohistochemistry may facilitate the identification of red cell precursors. Good results have also been achieved with rituximab plus corticosteroids, rituximab, cyclophosphamide and dexamethasone or rituximab, dexamethasone and ciclosporine. Splenectomy can be useful in individual cases impossible to control by other measures, but is less and less indicated. Current evidence suggests that intense immunosuppression with agents such as fludarabine and alemtuzumab may trigger transformation. The diagnosis can be reliably suspected on clinical grounds, but needs to be confirmed by biopsy. Patients respond badly to conventional lymphoma regimens and allogeneic stem cell transplantation should be considered in eligible patients. Skin cancers other than melanoma can be also observed, but at a frequency no different from that of the general population. It is usually considered that there is no relationship between treatment and the incidence and type of secondary solid tumours. The possibility of a second tumour should be entertained whenever a given patient presents with unexpected, unusual symptoms. Their pathogenesis is multifactorial, including hypogammaglobulinaemia, immunosuppression and treatment-related myelotoxicity. In some studies it has been found that infections are more related to prior therapy and diminished bone marrow reserve than to hypogammaglobulinemia. With chlorambucil, most infections are bacterial and frequently involve the respiratory tract. The pathogenesis of infections with purine analogues is related to T-cell abnormalities induced by these agents, with herpes virus infections being very frequent. Infections by Pneumocystis, Listeria, Mycobacteria, Aspergillus and Candida can be also observed. Patients with evidence of prior hepatitis B infection should be monitored for clinical and laboratory signs of reactivation during therapy and for several months after its completion.
Order viagra once a day
Less-severe manifestations (modified smallpox or variola sine eruptione) occurred in some vaccinated individuals doctor who cures erectile dysfunction generic viagra 50mg on line, whereas hemorrhagic or flat-pox types of smallpox occurred in patients with impaired immune responses. Variola major smallpox was differentiated into four main clinical types: (i) ordinary smallpox (90% of cases) produced viremia, fever, prostration, and rash; (ii) modified smallpox (5% of cases) produced a mild prodrome with few skin lesions in previously vaccinated people; (iii) flat smallpox (5% of cases) produced slowly developing focal lesions with generalized infection and an 50% fatality rate; and (iv) hemorrhagic smallpox (<1% of cases) induced bleeding into the skin and the mucous membranes and was invariably fatal within a week of onset. A discrete type of the ordinary form resulted from alastrim variola minor infection (10). Prior to its eradication, smallpox as a clinical entity was relatively easy to recognize, but other exanthematous illnesses were mistaken for this disease (10). For example, the rash of severe chicken pox, caused by varicella-zoster virus, was often misdiagnosed as that of smallpox. However, chicken pox produces a centripetally distributed rash and rarely appears on the palms and soles. In addition, in the case of chicken pox, prodromal fever and systemic manifestations are mild, if present at all. Chicken pox lesions are superficial in nature, and lesions in different developmental stages may be present on the same area of the body. Other diseases confused with vesicular-stage smallpox included monkeypox, generalized vaccinia, disseminated herpes zoster, disseminated herpes simplex virus infection, drug reactions (eruptions), erythema multiforme, enteroviral infections, scabies, insect bites, impetigo, and molluscum contagiosum. Monkeypox was first recognized by Von Magnus in Copenhagen in 1958 as an exanthem of primates in captivity. The clinical appearance of human monkeypox, typified by the Congo Basin variant, is much like that of smallpox, with fever, a centrifugally distributed vesiculopustular rash (appearing also on the palms and soles), respiratory distress, and, in some cases, death from systemic shock. Like variola virus, monkeypox virus appears to enter through skin abrasions or the mucosa of the upper respiratory tract, where it produces an enanthem and cough. During the primary viremia, the virus then migrates to regional lymph nodes, and during secondary viremia, it is disseminated throughout the body and the skin rash appears. Poxviruses n 1831 phadenopathy (generally inguinal) with fever and headache is common. Individual skin lesions develop through stages of macule, papule, vesicle, and pustule. Sequelae involve secondary infections, permanent scarring and pitting at the sites of the lesions, and sometimes alopecia and corneal opacities. Acute illness in the United States in 2003, caused by the "West African" variant, appeared generally more mild (19, 20); genomic sequence analyses and comparative epidemiologic and clinical data support the existence of two distinct clades of monkeypox virus. Additional information on clinical manifestations of disease (46) is also available. Generally, few lesions develop, and these occur primarily on the skin of the upper arms, face, neck, or trunk (50). Symptoms that occur prior to the appearance of lesions include fever, backache, and headache. Vaccinia Virus Humans have historically encountered vaccinia virus most commonly in the form of smallpox vaccine (now called vaccinia vaccine), a live-virus preparation that is crossprotective against other orthopoxvirus infections. Vaccination is done by using a multiple-puncture technique that causes a local lesion, which develops and recedes in a distinctive manner in primary vaccinees during a 3week period. At the site of percutaneous vaccination, a papule forms within 2 to 5 days, and the lesion reaches maximum size (1 cm in diameter) by 8 to 10 days postvaccination after evolving through vesicle and pustule stages; an areola may encircle the site. The pustule dries into a scab, which usually separates by 14 to 21 days after vaccination. In some vaccinated children, fevers with temperatures as high as about 100°F have occurred, though these have been uncommon in adults, and a regional lymphadenopathy has been observed. Despite attempts to prescreen potential vaccinees for contraindications, instances of generalized vaccinia rash, which may arise 10 to 14 days postvaccination, continue to be reported (48). On a clinical basis alone, it is often difficult to distinguish between generalized vaccinia, which represents virus presumably spread hematogenously, and a form of erythema multiforme, which may be immunologically mediated. Laboratory identification of virus within the disseminated rash may differentiate these conditions (48). Human orf virus infection is usually found on the fingers, hands, and arms but may also be found on the face and neck. Fever and swelling of draining lymph nodes may be present, and the lesions often ulcerate and are painful. Molluscipoxvirus In children and teenagers, molluscum contagiosum lesions generally appear on the trunk, limbs (except the palms and soles), and face, where there may be ocular involvement. Lesions are pearly, flesh-colored, raised, firm, umbilicated nodules, 5 mm in diameter. Cowpox Virus In humans, cowpox lesions occur mainly on the fingers, with reddening and swelling. Autoinoculation of other parts of the body may occur, and severe systemic infections have been reported. Antiviral Therapy Currently there are no drugs approved for use in the treatment of poxvirus infections; this is an area of active research and development (52). Additionally, laboratories testing samples from laboratory workers with potential occupational exposures to vaccinia virus may wish to consider vaccination of staff, in addition to the use of biosafety level 3 containment facilities, equipment, and work practices. Suitable specimens for laboratory testing of most suspected poxvirus infections are at least two to four scabs and/ or material from vesicular lesions. Scabs can be separated from the underlying intact skin with a scalpel or a 26-gauge needle, and each specimen should be stored in a separate container to avoid cross-contamination. Coexistent infectious rash illnesses, including simultaneous chicken pox and monkeypox infections, have been noted (14). Lesions should be sampled so that both the vesicle fluid and the overlying skin are collected. Once the overlying skin is lifted off and placed in a specimen container, the base of the vesicle should be vigorously swabbed with a wooden applicator or polyester or cotton swab. The viscous material can be applied onto a clean glass microscope slide and air dried. A "touch prep" can be prepared by pressing a clean slide onto the opened lesion by using a gradual pressing motion. If available, three electron microscope grids can be applied in succession (shiny side to the unroofed vesicle) to the lesion by using minimal, moderate, and mod- erate pressure. The glass slides and electron microscope grids should be allowed to air dry for about 10 min and then placed in a slide holder or a grid carrier box for transport to a laboratory. Alternative lesion sampling processes, including storing material on appropriate filter paper types, are being evaluated. Sample storage in transport medium (as done, for example, with herpesviruses) is discouraged, since specimen dilution decreases the sensitivity of direct evaluation by electron microscopy. A biopsy of lesions may also provide material suitable for direct viral evaluation or immunohistochemistry. A 3- to 4-mm punch biopsy specimen can be made, and the specimen can be bisected, with half placed in formalin for immunohistochemical testing and the remainder placed in a specimen collection container. Blood and throat swabs obtained from suspected smallpox patients during the prodromal febrile phase and early in the rash phase were also a potential source of virus during the smallpox era (56). Patient serum can also be obtained for serology to substantiate viral infection diagnoses or to infer a retrospective diagnosis. Paired acute- and convalescent-phase serum specimens can be of great value for diagnosis of infection. In this case, serum should be obtained as early as possible in the disease course and then 3 to 4 weeks later. Most virus-containing specimens should be stored frozen at -20°C or on dry ice until samples reach their transport destination. Electron microscopy grids and formalin-fixed tissues should be kept at room temperature. In addition, multiple different clinical laboratory tests can be useful for identifying and differentiating poxviruses, including electron microscopy, antigenic testing, nucleic acid detection, determination of virus growth features, and serology. The utility of these test methods for the diagnosis of poxvirus infections is shown in Table 2. Negative-stain electron microscopy of lesion samples was widely used during the smallpox eradication era. Poxviruses n 1833 hemadsorption or hemagglutination assays using chicken erythrocytes that are pretested to be suitable for such tests.
Buy generic viagra 25 mg
HbE is slightly unstable in vitro latest news erectile dysfunction treatment purchase viagra cheap online, although it is not clear how clinically significant this is. There are thalassaemic red cell changes, and the bone marrow shows marked erythroid hyperplasia. There is nearly always anaemia and splenomegaly, with typical thalassaemic bone changes. There are thalassaemic red cell changes and the bone marrow shows marked erythroid hyperplasia. Although relatively little is known about the natural history of this disorder, it is clear that in many parts of Southeast Asia and India it causes a very high mortality in early life. Clinically it is indistinguishable from other forms of -thalassaemia, although the link between genotype and phenotype is less predictable, with some patients having thalassaemia major and others, being non-transfusion dependent, growing and developing with few complications. The diagnosis is confirmed by finding only HbE and HbF on haemoglobin electrophoresis and by demonstrating HbE trait in one parent and -thalassaemia trait in the other. In cases of HbE/+ -thalassaemia, variable quantities of HbA are present and the condition is usually milder. Variant forms of -thalassaemia Despite the vast heterogeneity of mutations, the increased levels of HbA2 in -thalassaemia heterozygotes is remarkably uniform, in the range 3. The unusually high HbA2 levels are usually accompanied by higher than usual increases in HbF, resulting in a milder thalassaemia phenotype, despite the absence of HbA2 in some cases. Otherwise, the haematological picture is identical to the common forms of thalassaemia. Some -thalassaemia heterozygotes have normal HbA2 levels, despite the typical hypochromic microcytosis. Other cases of normal HbA2 -thalassaemia are extremely mild forms of -thalassaemia that is completely silent in heterozygotes and is only identified when it is coinherited with a common form of -thalassaemia. Heterozygotes do not have any evident Chapter 6 Haemoglobin and the inherited disorders of globin synthesis haematological phenotype, the only abnormality being a mild imbalance of globin chain synthesis. The clinical picture is characterized by a moderate degree of anaemia and splenomegaly with marked thalassaemic changes of the red cells, and ineffective erythropoiesis with intracellular inclusion bodies. Although not usually transfusion dependent, such individuals develop iron overload from hyperabsorption due to ineffective erythropoiesis and may develop liver or endocrine damage. Unlike the recessive forms that are prevalent in malarious regions, dominantly inherited -thalassaemias are rare, occurring in dispersed geographical regions. Most of the dominant -thalassaemia alleles have been described in single families, many as de novo mutations. Dominantly inherited thalassaemia should be suspected in any patient with a thalassaemia intermedia phenotype, even if both parents are haematologically normal, and the patient is from an ethnic background where -thalassaemia is rare. The distinction between them is subtle and originally made on what appeared to be clear-cut clinical and haematological grounds. However, as more cases became recognized and their underlying mutations delineated, it became evident that there is considerable overlap between the two groups of disorders. Compound heterozygotes of -thalassaemia with -thalassaemia, and -thalassaemia homozygotes have disease severity that ranges from mild anaemia to transfusion dependence. Heterozygotes for Hb Lepore have hypochromic microcytic red cells, normal HbA2 and variably increased HbF. Deletions that leave both the -globin genes intact are called G A ()0 -thalassaemias, and they all include parts or all of the - and -globin genes. G (A)0 -thalassaemia deletions include part or all of the A globin gene and hence produce HbF containing only G -chains. A couple of the ()0 -thalassaemias include two deletions separated by an inverted region. The mutations (single-base substitutions or minor deletions) are clustered in three regions of the promoters, around positions -114, -175 and -200. These regions contain binding sites for ubiquitous and erythroid-specific factors. Altered binding patterns of the transcription factors due to the point mutations are thought to be the cause of the elevated HbF levels, which vary from 5 to 35% in heterozygotes. Inherited increases in HbF have also been reported in families with -thalassaemia and sickle cell disease; these increases had a modulating effect on the severity of disease and the inheritance appeared to segregate independently of the -globin gene cluster. In African-American patients with sickle cell disease, the three loci contribute more than 20% to the HbF variation with a corresponding reduction in frequency of acute pain. Coinheritance of the Xmn1-G site delays transfusion requirements in -thalassaemia. Heterozygotes have severe haemolytic disease of the newborn, with anaemia and hyperbilirubinaemia. The severity of anaemia and haemolysis is variable, even within a family, and in some cases blood transfusions are necessary during the neonatal period. If they survive the neonatal period, the infants grow and develop normally; in adult life they have the haematological picture of heterozygous -thalassaemia, with mild anaemia, hypochromic microcytic red cells and a haemoglobin pattern of normal HbA2 -thalassaemia. Mediterranean region, the Middle East, the Indian subcontinent and Southeast Asia, in a line stretching from southern China through Thailand, the Malay Peninsula and Indonesia, to the Pacific Island populations. In some prevalent areas, carrier frequency for the mild form (+) reaches 80% or more. The more severe forms (0 thalassaemias) reach their highest frequency in Southeast Asia, where carrier frequency can reach 10%. The -thalassaemias can be classified as 0 -thalassaemia, in which no -chains are produced from the linked pair, and + thalassaemia, in which production of -chain from the affected chromosome is reduced. The majority of the non-deletional -thalassaemia (more than two-thirds) are found on the dominant 2 gene, less than one-third on the 1 gene and the others on an /-chromosome (/-T). In general, the non-deletional + -thalassaemia variants (/T) give rise to a more severe reduction in -chain output than the single -gene deletion (/-) due to the lack of compensatory increase in -output from the linked 1 gene, as observed in deleted cases. This results in an elongated -globin chain of 172 residues, 31 amino acids from the natural arginine at codon 141. Of the six predicted 2 -chain termination variants, five have been described: Hb Constant Spring (142 Gln), Hb Icaria (142 Lys), Hb Koya Dora (142 Ser), Hb Seal Rock (142 Glu) and Hb Pakse (142 Tyr). Hb Constant Spring is by far the most common of these variants, reaching frequencies of up to 4% in Thailand. Unusual causes of -thalassaemia this includes a single case report of 0 -thalassaemia arising from a deletion involving the 1 gene that also inactivated the intact linked 2 gene. Another novel form of non-deletional -thalassaemia results from a single nucleotide substitution in a non-genic region between the -globin genes and their upstream regulatory elements. The single-base substitution leads to the creation of a new promoter-like element that interferes with normal activation of all the downstream -like globin genes, resulting in 0 -thalassaemia. Pathophysiology the pathophysiology of -thalassaemia is different to that of thalassaemia. These soluble tetramers do not precipitate extensively in the bone marrow and hence erythropoiesis is more effective than in -thalassaemia. The inclusion bodies cause red cell membrane damage and obstruction in the spleen leading to shortened red cell survival. The molecular basis of the + -thalassaemias is more complicated; the commonest forms result from deletions that remove one of the linked pairs of -globin genes, leaving the other intact (-/). Less commonly, both the -globin genes are intact and + thalassaemia results from point mutations that partially or completely inactivate one of them (T /). Loss of two -genes (- -/-or -/-) produces a mild (a) 1 hypochromic microcytic anaemia, the -thalassaemia trait. As in -thalassaemia intermedia, HbH disease spans a wide range of clinical and haematological phenotypes, with equally heterogeneous genotypes, varying with the geographic distribution of the different -thalassaemia variants. Less often, it can result from the interaction of 0 -thalassaemia with non-deletional forms of -thalassaemia (/T) or from homozygous non-deletional thalassaemia (T /T). As the non-deletional forms of + -thalassaemia tend to have a more severe phenotype than the deletional forms, in some cases the homozygous state (T /T) may be associated with the phenotype of HbH disease.

Order viagra uk
In patients with sideroblastic anaemia erectile dysfunction protocol food lists cheap viagra 75 mg otc, the erythroblasts are often poorly haemoglobinized or show cytoplasmic vacuolation. The list of dysplastic features is considerable and may include binuclearity and multinuclearity, internuclear bridging, nuclear budding and fragmentation, increased pyknosis and basophilic stippling. At least 200 cells in the peripheral blood require evaluation and 10% of any lineage must be found to be dysplastic to count as evidence of dysplasia. There is commonly marked anisocytosis/poikilocytosis and the red cells tend to be macrocytic and oval-shaped. In sideroblastic anaemia, the blood film is classically dimorphic, containing a minority population of hypochromic microcytic cells; Pappenheimer bodies, which can be confirmed with an iron stain, and basophilic stippling may also be seen. Some patients have occasional circulating erythroblasts in the peripheral blood that are often dysplastic or megaloblastic. Granulocytic precursors may show cytoplasmic basophilia with aberrant granulation. Prominent basophilic and eosinophilic differentiation and increased numbers of blasts may be present. Megakaryopoiesis is commonly dysplastic, of which the most specific feature is the micromegakaryocyte. This is typically the size of a myeloblast with an ill-defined or blebbed outline and a single monolobed nucleus. Other megakaryocytes may exhibit hypolobulation or contain multiple disparate nuclei due to aberrant maturation. Bone marrow histology Histological analysis of the bone marrow can yield diagnostic information not apparent by aspirate morphology, particularly regarding bone marrow cellularity, architectural changes and stromal reactions. Cytological evidence of dysplasia can be found in all lineages, but is most easily detected in the erythroid precursors. However, detection of these provide evidence of clonality and are useful in patients with normal karyotypes. However, various abnormalities are discernible, notably low side-scatter, reduced expression of normal myeloid markers and aberrant patterns of expression of other markers. These include the following: 1 Granulocyte function tests to demonstrate defective phagocytosis, cell killing and motility. Cytogenetic analysis represents the most important investigation at diagnosis, both for understanding the biology of the disease and for making prognostic recommendations for the patient. Their analysis was based on a total of 24 different karyotypic abnormalities, ranked according to the median survival of patients with each abnormality, which were then grouped into four risk subgroups. However, normal karyotype accounted for just over 50% of all the cases studied and these could be categorized into the good-risk group. The biology of the disease, which dictates the rates of clonal expansion and leukaemic evolution, clearly involves genetic and epigenetic abnormalities. These patients were either untreated or had received only short courses of low-dose oral chemotherapy or haemopoietic growth factors. A global analysis was performed and critical prognostic variables were evaluated to generate a consensus prognostic scheme, particularly using more refined bone marrow cytogenetic information. In addition to patient age, univariate analysis Chapter 25 the myelodysplastic syndromes Table 25. The value of such a prognostic scoring system is clearly dependent on the type of treatment that a patient receives. Based on this score, patients are allocated into one of four risk groups: low, intermediate-1, intermediate-2 or high. The median survival (in months) of these five groups was 136 (very low), 63 (low), 44 (intermediate), 19 (high) and 8 (very high). The likelihood of developing leukaemia ranged from a 10-year probability of 7% in the very low-risk group to a 50% probability at 8 months in the very high-risk group. No doubt further revisions will emerge in the future that incorporate additional discriminatory risk factors. The karyotypic abnormalities and risk categories required more than 10 patients to be included as a specific category. Cytopenias are assessed individually with increasing risk for depth of individual cytopenias (Tables 25. Age-adjusted cut-offs are applied to put patients into five discrete prognostic groups: very low (median overall survival 8. The score has been validated in an independent cohort and prospectively in several countries, is of relevance in predicting outcomes in response to disease-modifying therapies, such as treatment with azacytidine and allogeneic stem cell transplantation. This work is being refined further in larger data sets to derive a molecular prognostic scoring system. Furthermore the number of acquired somatic mutations are also prognostic, with a poorer prognosis attached to increasing numbers of mutations. The management recommendations in this chapter are drawn partly from these guidelines, but have also sought to include newer treatment options that have emerged since the guidelines were published. Where appropriate consideration should be given to treatment with erythropoietin to alleviate anaemia, as discussed below. However, their long-term use, for instance in the prevention of recurrent epistaxis or oral bleeding in elderly patients with persistent thrombocytopenia, presents significant logistical issues. Moreover, as for red cell transfusions, platelet transfusions are not without potential complications, including allosensitization that can lead to refractoriness. Antifibrinolytic agents such as tranexamic acid can be useful on occasion, but are not routinely recommended. Where patients are likely to undergo intensive chemotherapy followed by transplantation, the use of leucodepleted products and irradiated products should be considered. In such patients, there is evidence that cytokines can act on haemopoietic progenitor cells to reduce apoptosis and improve erythropoiesis. Moreover, transfusion dependency or the surrogate Hb values of <90 g/L in males and <80 g/L in females predict an increased risk of cardiac comorbidity and reduced overall survival. Thus, patients who exhibit symptoms or signs of clinical anaemia should receive red cell transfusions in order to improve quality of life and ideally maintain Hb above these thresholds. For patients with coexisting cardiac dysfunction, anaemia may precipitate cardiac failure and individualized transfusion goals may help alleviate this. However, the potential risks of blood transfusions should always be considered, notably iron overload in multitransfused patients. Therefore, transfusions should only be used to alleviate symptoms of anaemia and not simply to maintain the haemoglobin above an arbitrary level. Other factors that might accentuate anaemia, such as nutritional 462 Chapter 25 the myelodysplastic syndromes Care must be taken not to exceed 120 g/L, due to the increased risk of thrombosis, which is approximately 2% particularly if there are coexisting vascular risk factors such as previous stroke, diabetes or hypertension. Similarly, darbepoietin is a recombinant long-acting erythropoietin that differs from the native form in having two additional N-linked oligosaccharide chains. Treatment with darbepoietin may be commenced at 300 g every 14 days, which may be increased after 8 weeks to 300 g per week for a further 8 weeks.
Purchase viagra overnight
As the tumor is large (>1cm) erectile dysfunction drugs from himalaya discount viagra uk, and there is axillary lymph node metastasis, postoperative 6 cycles chemotherapy is given with either of the two regimes: i. Male breast cancer is estrogen receptor positive in 70 to 80 percent cases and progesterone receptor positive in about 65 percent cases. Then the operation of modified radical mastectomy is done followed by adjuvant therapy. Postoperative radiotherapy to the breast flap and lymph node fields for locoregional control of the tumor. Then every 3 months for 12 months, then every 6 months for 12 months, then yearly for 10 years. Orchiopexy or orchidopexy is the operation of bringing down the testis and fixing it in the scrotal sac. The testis is brought into the subdertos pouch by making a small incision in the dartos muscle. Testis is brought out through the scrotum and is placed in the subcutaneous tissue of the inner side of thigh and is secured by stitches to fascia lata. Here the mobilized testis is placed in the opposite scrotum after making a small opening in the median septum. It is based on the principle that testicular artery is not an end artery and one of the factors hindering mobilization of testis is the short length of the testicular vessels. There is an expansile impulse on cough over the swelling and the swelling reduces easily on lying down. Testis is impalpable in the inguinal canal but there is an associated right inguinal hernia. When the descent of testis is arrested in some part of its pathway to scrotum, it is called imperfect descent or undescended testis. Testis is not palpable, location is established by combination of the following: (see also Q. A testis which has not descended by the end of first year will probably remain so. In this procedure testicular vessels are divided high up so that the testes could be mobilized well and brought down into the scrotum. The blood supply to the testis is maintained by artery to the vas and the cremasteric vessels. The testicular artery is anastomosed to inferior epigastric artery and testicular vein to inferior epigastric vein. Lockwood has explained location of ectopic testis by describing 5 tails of gubernaculum of testis viz. Superficial inguinal pouch which is a space between the external oblique aponeurosis and fascia of scarpa (Membranous layer of superficial fascia). In case of unitateral undescended testis, there may be subfertility due to defective spermatogenesis in the normally descended testis. Impalpable testis implies that the testis cannot be detected on physical examination. A detailed history and clinical examination including palpation along the route of normal descent and the ectopic sites. It is a type undescended testis near the superficial inguinal ring and may sometimes project beyond the ring on straining, but again slips back into the inguinal canal. How to differentiate between an ectopic testis in the superficial inguinal pouch and the canalicular testis The ectopic testis in the superficial inguinal pouch becomes more prominent as it is superficial to external oblique aponeurosis. The canalicular testis being deep to external oblique aponeurosis becomes less prominent. By the age of 4 years massive collagen deposition is evident and by the age of 16 years, irreversible destructive changes will occur. On examination, the swelling shows no expansile impulse on coughing and there is get above the swelling. This is a collection of serous fluid in the tunica vaginalis testis or any part of processus vaginalis. Primaryhydrocele:Herethetestisand epididymis are normal and the cause is not clear. Secondary hydrocele: Here the hydrocele is secondary to a disease of the testes and epididymis. Vaginal hydrocele-In this condition there is abnormal collection of serous fluid between the visceral and parietal layers of tunica vaginalis. Infantile-When processus vaginals is obliterated at the deep inguinal ring and accumulation of fluid takes place upto that level, it is called infantile hydrocele. Congenital hydrocele-When processus vaginalis communicates with the peritoneal cavity and fluid accumulates in it, it is called congenital hydrocele. Encysted hydrocele of cord-When only a small portion of processus vaginalis remains patent, it is called encysted hydrocele of cord. Steady pressure is applied at the lower pole with the thumb and fingers of the other hand. Observation: Thumb and fingers at the upper pole are passively raised and / or appreciably separated. It is a condition in which there is a collection of fluid in the hernial sac, after the neck of the sac gets closed by a tag of omentum. It is the hydrocele developed following herniorrhaphy due to lymphatic injury in the cord. It causes inguinoscrotal swelling but one can get above the swelling and there is no expansile impulse on coughing. In congenital hydrocele, fluid comes from the peritoneal cavity but in vaginal hydrocele; fluid is secreted by epithelial covering of the tunica vaginalis testis. The treatment of congenital hydrocele is the same as congenital hernia that is, herniotomy through an inguinal approach. The light should not be thrown from the back of the swelling as the testis will come on the way and the test will be negative. The upper pole of the swelling is held between the thumb and forefingers of one hand to make the swelling tense and steady. The swelling has got free mobility but when traction is applied to testis gently, the swelling becomes fixed. In this condition, the processus vaginals remains patent in the middle being shut off from the tunica vaginals below and peritoneum above. In case of encysted hydrocele of cord, when traction is applied to testis gently, the swelling becomes fixed as it is related to the cord. It resembles encysted hydrocele but occurs in female in relation to round ligament. Testicular vein is dissected in the retroperitoneum lateral to the external iliac artery. This is a triangular area bounded by the testicular vessels laterally and the vas deferens medially and the line joining these two structures above. This triangle is considered as dangerous for laparoscopic approach for varicocele or hernia because the external iliac vessels lie on its floor, covered only by peritoneum and the transversalis fascia. Hence sharp instrument dissection and application of electrocautery is dangerous in this triangle. Varicocele case summary the 22-year-old male patient presents with a swelling in the left side of scrotum for last 5 years. The swelling disappears on lying down position but reappears on standing and walking. On palpation there is a mass of dilated veins which feels like the bag of worms, on the left side of scrotum. Primary varicocele In more than 90 percent cases no cause is found and the varicocele is called the primary varicocele.
