

Order cialis soft 40 mg on-line
The adrenal gland contains specific cytochromes P450 involved in the biosynthesis and regulation of steroid hormones but also has the capacity for xenobiotic metabolism (Gustafsson et al impotence lotion order cialis soft 20mg visa. Most other tissues have lower specific contents of xenobiotic metabolizing enzymes but nonetheless have a limited capability to bioactivate and detoxicate chemical toxicants. Cytochromes P450, glutathione S-transferases, epoxide hydrolase, glucuronosyl transferase, and other enzymes involved in xenobiotic metabolism are readily inducible by a variety of substances (Conney, 1967). Exposure to many common environmental chemicals can induce xenobiotic metabolism, including pollutants, cigarette smoke, and dietary constituents. Enzyme induction in humans is well documented in the clinical setting (Conney, 1967). In general, enzyme induction increases the apparent Vmax for a biotransformation reaction because the total enzyme involved in the reaction increases. This can lead to increases or decreases in the biological effects of chemicals, depending upon whether the parent chemical or the metabolite is the active entity. As discussed earlier, two substrates for the same enzyme can act as competitive inhibitors toward each other. This sort of inhibition is reversible, and in time both chemicals will be metabolized and eliminated. As is the case with inducers, many potential inhibitors of xenobiotic biotransformation are present in the environment. The situation frequently encountered with multidrug therapy is enzyme induction and inhibition by the same agents. For example, ethanol inhibits drug metabolism upon acute administration but will induce cytochromes P450 and other enzymes upon chronic administration (Lieber et al. Cruciferous vegetables and charcoal-broiled meats contain potent enzyme inducers, and diets containing these foods have been shown to modulate Toxicokinetics: Biotransformation of Toxicants 139 the biological effects of xenobiotics (Conney et al. Nutrition also plays an important role in maintaining xenobioticmetabolizing enzymes and their cofactors (Parke and Ioannides, 1981). Most vitamins are involved in the production of cofactors required for xenobiotic metabolism. Dietary protein and lipid can also influence the enzymes involved in xenobiotic metabolism (Parke and Ioannides, 1981). There are three primary factors involved in the alteration of xenobiotic metabolism by liver disease. Changes in liver blood flow affect the delivery of the xenobiotic to the site of metabolism. A reduction in metabolic capacity is likely to occur in liver disease due to a diminished number of viable hepatocytes. Additionally, albumin production is frequently diminished in patients with liver disease, potentially resulting in a higher concentration of unbound drug. This can lead to higher tissue concentrations of the drug and a greater potential for toxicity (Howden et al. Other disease states such as diabetes and hypertension can also lead to changes in xenobiotic metabolism (Schenkman et al. Stress has also been shown to produce changes in xenobiotic metabolism and toxicity (Vogel, 1993). The alcohol dehydrogenase-dependent metabolism of allyl alcohol in rats increases with age, leading to an age-dependent increase in hepatotoxicity. However, the cytochrome P450-dependent bioactivation of acetaminophen and carbon tetrachloride decreases with age (Rikans, 1989). Aging also affects physiological factors such as fat content and blood flow that can affect xenobiotic metabolism. These sex differences in xenobiotic metabolism are controlled by the hypothalamus and pituitary gland (Gustafsson et al. Sex differences have also been observed in other species, including humans (Anderson, 2008), but have not been as well studied. In general, gender differences in xenobiotic metabolism are more pronounced in rodents than in humans. Extensive polymorphisms have been described for cytochromes P450 1A1, 1A2, 2A6, 2A13, 2B6, 2C8, 2C9, 2C19, 3A4, 3A5, and 2D6. The polymorphism in cytochrome P450 2D6 is of great clinical importance since this enzyme metabolizes over 30 different therapeutic agents. The distribution of the variant alleles of cytochrome P450 genes varies among different ethnic populations (Zhou et al. A polymorphism in flavin-containing monooxygenase form 3 has been described for the N-oxidation of dietary trimethylamine, called trimethylaminuria or fish-odor syndrome (Cashman and Zhang, 2002). Polymorphisms have been described in several alcohol dehydrogenase genes that may explain the interethnic variations observed in ethanol metabolism (Hoog and Ostberg, 2011). Polymorphisms in cytosolic epoxide hydrolases have been identified that may influence the metabolism of exogenous and endogenous epoxide substrates (Przybyla-Zawislak et al. Three classes of genetic polymorphisms in the glucuronidation of bilirubin have been described. High interindividual variations in hepatic and extrahepatic glucuronosyl transferases are influenced by single-nucleotide polymorphisms, regulation by transcription factors, and nuclear receptors (Bock, 2010; Hu et al. Single-nucleotide polymorphisms in sulfotransferases genes vary among different ethnic 140 Toxicokinetics: Biotransformation of Toxicants populations (Nowell and Falany, 2006; Lindsay et al. Polymorphisms in glutathione S-transferase genes are associated with increased susceptibility to carcinogenesis and inflammatory diseases (Hayes et al. The polymorphism occurs in approximately 50% of Europeans and as many as 90% of Moroccans resulting in slow acetylation. This polymorphism is associated with several drug-induced toxicities, including lupus erythematosus. It has also been associated with increased occupationally induced bladder cancer in slow acetylators and increased colon cancer in rapid acetylators (Daly et al. Thiopurine S-methyltransferase exhibits a polymorphism in 10% of Caucasians, with 1 in 300 individuals having a complete deficiency. Patients with compromised thiopurine S-methyltransferase activity are at risk for toxicity after standard doses of thiopurine medications, including fatal myelosuppression (McLeod and Siva, 2002; Zhou, 2006). The biotransformation of toxicants is catalyzed by enzymes that have various kinetic mechanisms of combining with substrates and releasing products. Understanding the kinetic mechanisms of the enzymes involved in xenobiotic biotransformation is important for understanding mechanisms of toxicity and designing antidotal therapy. Exposure to multiple chemicals can lead to exaggerated biological effects due to modulation of the metabolism of one toxicant by another. These modulations generally occur by inhibition of biotransformation or by induction of enzyme activity. Characterization of the mechanisms of inhibition of biotransformation provides the basis for therapeutic intervention. Understanding the relationship of chemical biotransformation to adverse biological effects is often times central to understanding mechanisms of toxicity. The initial biotransformation of most organic chemicals (phase I metabolism) is catalyzed by the cytochrome P450 enzymes of the endoplasmic reticulum of liver cells. This superfamily of enzymes oxidizes and reduces carbon and a number of heteroatoms. The biosynthesis of many isoforms of cytochrome P450 is readily induced by exposure to various chemical agents. Flavincontaining monooxygenase is also important in the metabolism of nitrogen and sulfur compounds. Typical conjugation reactions involve glutathione, glucuronic acid, sulfate, and amino acids. While conjugation reactions usually represent detoxication, they can also result in the formation of reactive, toxic species. Although the majority of xenobiotic biotransformation takes place in the liver, most organs have some capacity for xenobiotic metabolism.
Purchase cialis soft discount
Acetaminophen-induced liver injury is also caused by a chemically reactive metabolite erectile dysfunction treatment sydney discount cialis soft 40mg overnight delivery. The formation of this metabolite occurs at a very low level after subtoxic doses, but increases as the dose approaches the toxic range. Endogenous compounds such as glutathione, a low molecular weight tripeptide found in cells, play an essential role in protecting liver cells from injury from chemically reactive intermediates. Overdoses of drugs such as amitriptyline, estradiol, and diazepam can cause a diminution or cessation of bile flow. Inflammation or blockage of the bile ducts can result in the retention of bile salts, or cholestasis. Cirrhosis is a progressive disease caused by the accumulation of collagen in the liver, typically due to chronic consumption of ethanol. However, a type of chemical- Table 4 Some hepatotoxicants and their associated types of liver injury Compound Acetaminophen Bromobenzene Chloroform Carbon tetrachloride Thioacetamide Chloroform Carbon tetrachloride Ethanol Puromycin Tetracycline Amitriptyline Imipramine Sulfanilamide Colchicine Halothane Phenylbutazone Zoxazolamine Aflatoxin B1 Pyrrolizidine alkaloids Urethane Vinyl chloride Type of injury Necrosis Fatty liver Cholestasis Hepatitis Cancer Source: From Plaa, G. Although a wide variety of chemicals have been shown to cause liver cancer in experimental animals, only a few are known to be human carcinogens. Two known primary human liver carcinogens are vinyl chloride and the mycotoxin aflatoxin B1. Furthermore, the kidney produces a number of critical hormones that influence metabolic functions. Accordingly, a toxicological insult to the kidney can have an impact on any of these functions. The kidney is particularly sensitive to the toxic effects of a variety of chemicals, primarily because of its unique anatomy and physiological features. For example, the extensive filtering and reabsorptive capabilities of the kidney cause remaining materials to be concentrated. Thus, a nontoxic concentration of a chemical in the plasma could become toxic in the kidney as the urinary filtrate is concentrated to form urine. Although the two kidneys comprise less than 1% of the total body mass, they receive approximately 25% of the cardiac output. Due to the high blood flow to the kidneys, any toxicant that is present in the systemic circulation will be delivered to the kidney in significant amounts. A number of chemicals found commonly in the environment may be toxic to the kidney (nephrotoxicity). For example, many metals, such as mercury and cadmium, are potent nephrotoxicants. At low doses, a variety of metals may cause alterations in ion transport capacity (aminoaciduria or glucosuria), whereas higher exposure can cause kidney cell necrosis and death. Extensive data has accumulated on the nephrotoxicity of mercury; the potential for nephrotoxicity of this compound is highly dependent upon its chemical form. The kidney is a primary target of toxicity following accidental or suicidal ingestion of mercuric salts. Thus, low levels of chronic exposure will eventually result in accumulation to toxic levels. Kidney damage has also been observed following administration of chromium, arsenic, gold, lead, and thallium. Many chlorinated hydrocarbons such as chloroform and hexachlorobutadiene also cause renal toxicity. In the case of chloroform, nephrotoxicity is somewhat dependent upon bioactivation to a toxic intermediate. Interestingly, the nephrotoxicity of several of the halogenated hydrocarbons may be related to the activation in the kidney of a conjugation product between the toxicant and an endogenous compound that is formed in the liver. Certain antibiotics are nephrotoxicants in humans when present in high doses or over prolonged periods of time. In particular, the aminoglycoside antibiotics, including streptomycin, neomycin, and gentamicin can cause kidney damage after prolonged use. The immunosuppressant drug, tacrolimus, and similar "calcinurin inhibitor" drugs used to reduce organ rejection in transplant patients are nephrotoxic in a significant percentage of transplant patients and may cause complete renal failure. Since the lung receives all of the cardiac blood supply, the distribution of inhaled toxicants from the lung to other organs can be rapid. Thus, it is important to distinguish between inhalation toxicology, which defines the route of exposure, and pulmonary toxicology, which specifically assesses the response of the lung to toxic agents. The lung is in direct contact with the external environment and is exposed to infectious agents as well as toxic particles and gases. Since the primary purpose of the respiratory system involves the exchange of gases, impairment of this process may affect the functions of the entire body, depending upon the degree of severity of damage. Over 40 different cell types are required to perform the diverse functions of the respiratory tract. In response to toxicant exposure, many of these lung cells are known to release a variety of chemical mediators that are designed to neutralize or remove the inhaled toxic material. The type of response mounted by the lung ultimately depends upon the physical and chemical properties of the agent. Some toxicants may elicit nonspecific responses involving clearance of the toxicant. Unlike most organs, the lung can respond to a toxic insult or agent by initially trying to remove or neutralize it and then repair the damage. These nonspecific responses provide a considerable degree of protection against injury from a wide variety of inhaled agents. In contrast, specific defense mechanisms are immunological in nature and are stimulated by the constant exposure to inhaled toxic antigens. Once sensitized to a particular antigen, the immune system can mount an amplified response to extremely small concentrations of that toxic antigen. Despite the specific and nonspecific defenses of the lung, chronic injury to the lung as a result of toxicant exposure occurs all too often. Chronic lung injury occurs when the defenses and repair processes of the lung simply cannot cope with the damage resulting from either high levels of acute toxicant exposure or repeated exposure to low levels of the material. The result of the struggle between repair and injury can produce a wide range of pulmonary responses including fibrotic diseases, obstructive pulmonary diseases, and cancer. A number of workplace toxicants induce inflammatory processes at concentrations sufficient to cause fibrosis after chronic exposure. In particular, silicosis is a common fibrotic disease that occurs after chronic occupational exposure to crystalline silica. One major obstructive disease that can be caused by pollutant exposure is emphysema. Emphysema is characterized by the destruction of certain airspaces of the lung, resulting in a steady progression of functional disability. Emphysema is clearly General Overview of Toxicology 21 associated with heavy cigarette smoking and occurs late in life. In general, the contribution of occupational and environmental agents toward lung disease is overshadowed by damage attributed to cigarette smoke. The brain, spinal cord, and peripheral nerves are covered with a lining of specialized cells that restrict entry of molecules from adjacent tissue. The principal basis for this barrier is the tight junction that exists between endothelial cells of the nervous system. Despite this barrier, certain toxicants, including methyl mercury, trimethyltin, organophosphorous insecticides, and carbon disulfide, are specific for cells of the nervous system and result in serious nervous system impairment, often leading to death, when exposure is severe enough. The effects of neurotoxicants are typically classified based upon their site of action (Costa, 2013). Certain toxicants are specific for neurons, the principal cells of the nervous system. Examples of compounds that are associated with neuronal injury include methyl mercury, trimethyltin, and carbon disulfide. Myelin provides the electrical insulation of nerve cells, and its loss leads to a slowing of electrical impulses along nerve cells, or myelinopathy.
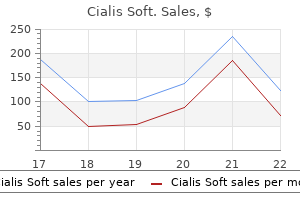
Buy 20 mg cialis soft fast delivery
The F344 rats are more sensitive than S-D rats to 1 erectile dysfunction drugs mechanism of action discount 40mg cialis soft visa,2-dichlorobenzene-induced hepatotoxicity. F344 rats were found to have a higher rate of tissue repair than that seen in S-D rats. Thus, despite much higher liver injury in F344 rats, presumably because of much higher bioactivation of 1,2dichlorobenzene, these rats escape the lethal consequence by very high rate of liver tissue repair, thereby equalizing lethality in these strains. This differential in tissue repair in the two strains plays a vital role in equalizing lethality, the ultimate outcome of toxicity in the two strains, despite dramatic difference in the 1,2-dichlorobenzene-induced hepatotoxicity. This results in complete recovery of the female rats even after much higher liver injury than male rats (Moghaddam et al. These gender differences may be explained by differences in bioactivation and tissue repair mechanisms. Regeneration impairment in hepatic miR-17 92-deficient livers was restored by ovariectomy, further supporting the role for estrogen signaling in regeneration. Similarly, various components of the diet affect liver regeneration either positively or negatively. High blood glucose inhibits liver regeneration while feeding lipid emulsions promotes liver regeneration. Administration of a standard amino acid mixture without energy substrate inhibits liver regeneration, whereas branched-chain amino acids such as valine, leucine, and isoleucine as well as glutamine promote liver regeneration (Chanda and Mehendale, 1996c; Holecek, 1999). The effects of other vitamins, micronutrients, and trace elements on liver regeneration after chemical-induced injury remain to be investigated. An important factor in facilitating compensatory liver regeneration after hepatic injury is the availability of cellular energy. The administration of nutritional substances to patients following liver damage is usually considered only with the aim of providing an energy source, without considering whether the nature of the energy substrate also influences liver regeneration (Zivny and Simek, 1989b; Zivny and Simek, 1989a). Glucose is used as a source of energy in patients with liver damage to treat marked hypoglycemia that often follows severe liver damage. Conversely, fatty acids and monoacetin facilitate tissue repair in the livers of animals. In contrast, rats fed with palmitic acid along with its mitochondrial carrier L-carnitine exhibited enhanced liver regeneration following thioacetamide treatment. Extensive studies with rats and mice have shown that moderate (35%) calorie restriction enhances liver regeneration following thioacetamide-induced liver injury (Ramaiah et al. Calorie restriction increases the signaling capability of the hepatocytes, resulting in a very rapid proliferation response compared to ad libitum-fed animals following liver injury. These data indicate that during calorie restriction, hepatocytes are maintained at an enhanced signaling state, which allows the cells to rapidly respond to tissue injury. The increased sensitivity and ability to respond to tissue injury in calorie restriction is reflected in the hepatic gene expression patterns of calorie-restricted animals (Apte et al. This increased sensitivity to drug-induced liver injury is due mainly to the decreased ability of the diabetic rats to initiate compensatory liver regeneration following toxicity. Diabetic rats have impaired cell cycle, resulting in blockage of G1-to-S phase transition. These data clearly indicate that the mechanism behind the increased susceptibility to chemical-induced liver injury in diabetes is related to cell cycle blockage induced by hyperglycemia resulting in decreased cell proliferation (Sawant et al. The primary mechanism behind this species difference in susceptibility to the chemicals is due to differences in liver regeneration and tissue repair in diabetic rats versus diabetic mice. Instead, the diabetic mice demonstrate rapid activation of mitogenic signaling, resulting in a robust liver regeneration (Shankar et al. Thus, the differential susceptibility to hepatotoxicants in diabetic rats versus diabetic mice is governed by the degree of liver regeneration and tissue repair in these species and provides additional evidence for the decisive role of tissue repair in the final outcome (survival vs. Evolutionary pressure has entrained the liver to adjust the balance between stimulators and inhibitors, while prevailing to meet its needs and the demands of the organism. Liver regeneration and tissue repair as an adaptive strategy are important both as a response to toxic insult and as a survival strategy from an evolutionary perspective. Work with a number of experimental hepatotoxicants indicates that exposure to toxic chemicals leads to two responses. Second, a simultaneous but opposing tissue repair response for recovery from injury is either stimulated, inhibited, or remains unaltered upon exposure to individual or mixture of chemicals depending upon the dose and other conditions of exposure. When tissue repair is inhibited, even an ordinarily inconsequential level of tissue injury may lead to fulminating liver failure and animal death from even a nonlethal exposure to hepatotoxicants. When tissue repair is unperturbed, the outcome may be mainly dependent on bioactivation and related mechanisms responsible for initiation of injury. When tissue repair is augmented, tissue injury becomes inconsequential to animal survival even though much higher tissue injury might be inflicted. Studies with interactive models of toxicity using a binary mixture of chemicals reveal that examples of each of the aforementioned sequels can be found. Findings from other models of chemical interactions such as autoprotection and heteroprotection underscore the importance of stimulated tissue repair in the final outcome of toxic injury. These findings suggest that animal survival, even after lethal overdose, is possible due to augmented tissue repair by a priming low dose of a toxicant. First, new cells are available for restoration of tissue structure and function by replacing the dead or dying cells. Second, the new cells are resilient to the action of toxicants, and this plays a significant role in restraining continued expansion of the initiated injury. Because toxicant-induced injury and stimulated tissue repair are simultaneous but opposing responses, measuring both of these responses in a dose-response paradigm is likely to increase the precision of predictive toxicology. One significant deficiency of the physiologically based pharmacokinetic models used in the mid-1990s is the lack of consideration of the "toxicodynamic" response of the body that initiates compensatory cell division after exposure to drugs and toxicants. Although some effort is being directed to include the biological response of stimulated tissue repair (el-Masri et al. Since factors such as exposure dose, age, gender, macronutrients, and so on are known to influence the tissue repair status and hence the ultimate outcome of toxicity, consideration of such quantitative relationships is likely to be helpful in accounting for interindividual variability in toxicity. Furthermore, species/strain differences in sensitivity to toxicants might be explained by differences in the tissue repair response elicited upon exposure to toxicants. The findings that the repair response can be manipulated and that such manipulation can lead to a complete reversal in the outcome of toxic injury open up new avenues for therapeutic applications in the realm of biomedicine. For instance, if availability of cellular energy is indeed the predominant reason behind failed biological response, therapeutic intervention by externally supplying cellular energy would be indicated. Death usually occurs or injury is prolonged when any of the recovery steps are inhibited (Mehendale, 2005). With the advent of gene therapy, specific genes could, one day, be delivered directly to the liver to induce the expression/suppression of desired factors implicated in liver regeneration. Studies on the age-dependent effects of galactosamine in primary rat hepatocyte cultures. Ongoing hepatocellular regeneration and resiliency toward galactosamine hepatotoxicity. Acquired resistance to acetaminophen hepatotoxicity is associated with induction of multidrug resistance-associated protein 4 (Mrp4) in proliferating hepatocytes. Delayed liver regeneration in peroxisome proliferator-activated receptor-alpha-null mice. Upregulated promitogenic signaling via cytokines and growth factors: potential mechanism of robust liver tissue repair in calorie-restricted rats upon toxic challenge. Mechanisms of increased liver tissue repair and survival in diet-restricted rats treated with equitoxic doses of thioacetamide. Enhanced liver regeneration following changes induced by hepatocyte-specific genetic ablation of integrin-linked kinase. Hepatic Defenses Against Toxicity: Liver Regeneration and Tissue Repair 391 Attisano, L. Inhibition of cyclooxygenase-2 aggravates secretory phospholipase A2-mediated progression of acute liver injury. Secretory phospholipase A2 mediates progression of acute liver injury in the absence of sufficient cyclooxygenase-2. Role of bile acids in liver injury and regeneration following acetaminophen overdose. Liver-specific deletion of integrin-linked kinase in mice attenuates hepatotoxicity and improves liver regeneration after acetaminophen overdose. Dual role of epidermal growth factor receptor in liver injury and regeneration after acetaminophen overdose in mice.
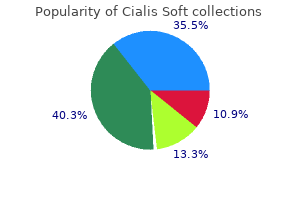
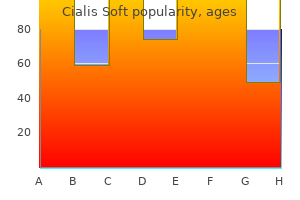
Order cialis soft 20 mg overnight delivery
As defense strategy to prevent the initiation or propagation of radical chain reactions best erectile dysfunction doctors nyc generic 20 mg cialis soft otc, these metals are transported and stored bound to proteins. It is a 24-subunit protein with a central core where iron is stored as ferric oxohydroxide (Theil, 2003, 2007). Two isoforms with heavy (H) or light (L) subunits are found in various tissues; the liver contains mainly the L-form, which is the dominant form for cells where iron is stored (Worwood, 1990). After iron administration, there is an increase of transcription of the L-gene; however, the main regulation of ferritin synthesis is under translational control (Worwood, 1990). Thus, oxidant stress may facilitate iron sequestration into ferritin and limit the detrimental effect of iron (Bouton, 1999; Fillebeen and Pantopoulos, 2002). One ferritin molecule can store up to 4500 atoms of iron; however, the protein is normally only 20% saturated (Harrison and Arosio, 1996). This suggests that iron has to be released from ferritin, transferrin, or lactoferrin to participate in Fenton reactions. In contrast, for iron to be released from a binding protein it has to be reduced (Minotti, 1993; Reif, 1992). In most cases, the experimental evidence involves the protective effect of an iron chelator, for example, desferoxamine, in the pathophysiology. Although its main biological function is the chelation and detoxification of metal ions (Klaassen et al. The acute-phase protein ceruloplasmin, which contains 95% of copper found in plasma, has no relevant function in copper transport or metabolism (Healy and Tipton, 2007; Hellman and Gitlin, 2002). Transferrin and lactoferrin can bind iron with high affinity and therefore virtually eliminate free iron from plasma. Haptoglobin/hemopexin in plasma binds heme/hemoglobin and prevents the release of redox-active iron (Gueye et al. It is important in protecting plasma lipoproteins against oxidation (Halliwell and Gutteridge, 1990). Uric acid is a direct scavenger of oxygen radicals and can also tightly bind iron and copper (Sevanian et al. It is an important antioxidant in the extracellular space due to its direct scavenging properties for a number of oxygen-derived free radicals and its capacity to regenerate a-tocopherol (Buettner and Jurkiewicz, 1996; Halliwell and Gutteridge, 1990). Its prooxidant activity in the presence of iron or copper ions is less relevant in vivo because transition metal ions are bound effectively. However, in certain disease states, for example, iron overload, high plasma levels of ascorbate may be detrimental (Buettner and Jurkiewicz, 1996; Halliwell and Gutteridge, 1990). Bilirubin bound to serum albumin may protect unsaturated fatty acids from peroxidation (Stocker et al. This enables the enzyme to bind to the proteoglycan, heparan sulfate in the glycocalyx of cell surfaces and connective tissue (Nozik-Grayck et al. Another potentially important antioxidant defense mechanism is hepatocellular release of the glycoprotein selenoprotein P (Burk and Hill, 2005). Because of its rapid turnover, $ 25% of the total-body selenium content passes through selenoprotein P per day (Burk and Hill, 2005). In addition to its 10 selenocysteine residues per molecule, it also contains 17 cysteines (Read et al. Selenoprotein P plays an important role in selenium homeostasis and can function as an antioxidant in the interstitial space (Burk and Hill, 2005). These data suggest a significant role of selenoprotein P as an antioxidant in the vascular space of the liver. As outlined earlier, cells have developed a highly effective, multitier system for the safe elimination of these reactive intermediates. The fact that impairment or elimination of individual components of the system causes problems ranging from enhanced susceptibility to oxidant stress to lethality indicates the vital importance of these systems. However, it is controversial whether boosting the antioxidant defense systems beyond their physiological levels can prevent acute or chronic disease processes. There are a number of reasons for these contradictory findings, including issues with the relevance of models for human diseases. This requires in-depth studies on the mechanisms of disease and the mechanisms of antioxidant action. Recently, an increased emphasis is being placed on the use of in vitro systems especially involving human cell types. It is important to be aware that the standard cell culture conditions use room air, which means that cultured cells are exposed to higher oxygen concentrations than cells in vivo. As a result, cultured cells are exposed to higher basic oxidant stress levels with modified gene expression profiles including those of antioxidant genes (Boess et al. These gene expression changes need to be considered because they can impact cell signaling pathways and affect mechanisms of cell injury and cell death and the extrapolation of the results to the in vivo systems. This is relevant to human pathophysiology, especially in the liver, since the organ receives both highly oxygenated blood from the hepatic artery as well as oxygen-depleted blood from the portal vein. FoxO transcription factors support oxidative stress resistance in human chondrocytes. The relationship of biliary glutathione disulfide efflux and intracellular glutathione disulfide content in perfused rat liver. Role of xanthine oxidase in small bowel mucosal dysfunction after surgical stress. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes: protection by N-acetyl cysteine. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Preferential transport of glutathione versus glutathione disulfide in rat liver microsomal vesicles. A major fraction of endoplasmic reticulum-located glutathione is present as mixed disulfides with protein. Superoxide anion generation in the liver during the early stage of endotoxemia in rats. Effects of hypochlorous acid and chloramines on vascular resistance, cell integrity, and biliary glutathione disulfide in the perfused rat liver: modulation by glutathione. Prevention of Kupffer cell-induced oxidant injury in rat liver by atrial natriuretic peptide. Glutathione protects the rat liver against reperfusion injury after hypothermic preservation. Glutathione treatment protects the rat liver against injury after warm ischemia and Kupffer cell activation. Inhibition of in vivo myocardial ischemic and reperfusion injury by a synthetic manganese-based superoxide dismutase mimetic. Gene expression in two hepatic cell lines, cultured primary hepatocytes, and liver slices compared to the in vivo liver gene expression in rats: possible implications for toxicogenomics use of in vitro systems. Selenoprotein P: an extracellular protein with unique physical characteristics and a role in selenium homeostasis. Pathogenesis of diquat-induced liver necrosis in selenium-deficient rats: assessment of the roles of lipid peroxidation and selenoprotein P. Fortilin potentiates the peroxidase activity of peroxiredoxin-1 and protects against alcohol-induced liver damage in mice. Protection against acetaminophen hepatotoxicity by clofibrate pretreatment: role of catalase induction. Oxidative stress promotes peroxiredoxin hyperoxidation and attenuates pro-survival signalling in aging chondrocytes. Neutrophil interaction with the hemostatic system contributes to liver injury in rats cotreated with lipopolysaccharide and ranitidine.
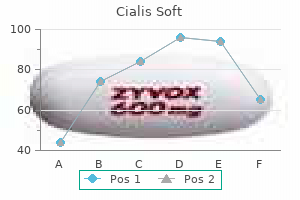
Order line cialis soft
MiR-146a impotence pump medicare buy cialis soft 40mg with amex, which is downregulated by lead exposure, is important for innate immune response in controlling the Toll-like receptor and cytokine signaling through a negative feedback regulation loop (Taganov et al. Humans are primarily exposed to mercury by consuming contaminated seafood or through occupational exposure (dentistry, mining, coal burning, and electric industries) (Boening, 2000; Tchounwou et al. There are three forms of mercury: elemental mercury, inorganic mercury compounds, and organic mercury compounds. Inorganic mercury compounds are water soluble and accumulate mainly in the kidneys, causing kidney damage (Park and Zheng, 2012). Organic mercury compounds (such as methyl and ethyl mercury) are 432 Epigenetics in Toxicology highly lipophilic and bioaccumulate in fish tissues, making them much more prevalent in human exposures. Mercury exposure has been associated with various adverse outcomes in neurological, cardiovascular, respiratory, and renal systems, as well as immune system (Tchounwou et al. Perinatal exposure to methylmercury causes persistent changes in learning and motivational behavior in mice (Onishchenko et al. Further, mouse embryonic stem cells exposed to mercury chloride have decreased total histone protein and decreased H3K27me1 (Gadhia et al. Exposure to nickel may occur through the inhalation of air, the ingestion of food and water, or skin contact (Sutherland and Costa, 2003). Occupational exposure to nickel has been associated with an increased risk of nasal and lung cancers in humans (1990; Beveridge et al. It has been proposed that nickel may act as an epigenetic carcinogen by structural changes of heterochromatin, which is associated with gene repression. In that study, all nickelexposed mice developed sarcomas at the implantation site, and hypermethylation in the promoter region of p16 was detected in the tumors. Nickel compounds also inhibit histone H4 acetylation in both yeast and mammalian cells, affecting only lysine 12 in mammalian cells and all of the four lysine residues (K5, K8, K12, and K16) in yeast (Broday et al. In epidemiological studies, H3K4me3 is elevated in peripheral blood mononuclear cells of Chinese subjects with occupational exposure to nickel, whereas H3K9me2 is decreased. Moreover, urinary concentrations of nickel have been positively associated with H3K4me3 (Arita et al. Human lung adenocarcinoma cells treated with nickel had increased H3K4me3 in both the promoters and the coding regions of several genes, as well as increased gene expression. This effect was accompanied by the crosslinking of chromatin in the coding regions immediately downstream of the transcription start sites of some nickel-induced genes (Tchou-Wong et al. Human exposure to phthalate occurs through ingestion, inhalation, and dermal contact. Exposure to BaP also causes hypomethylation events at a number of genomic repeat elements (Sadikovic and Rodenhiser, 2006). Epidemiological studies of air pollution have provided evidence of its adverse health effects, including asthma, chronic respiratory diseases, cardiovascular diseases, diabetes, and neurodevelopmental disorders (Samet et al. Additionally, lower methylation of the inducible nitric oxide synthase promoter has been observed in postexposure blood samples from steel plant workers (Tarantini et al. Exposure to cigarette smoke can cause various adverse health effects, including cardiovascular disease, cancer, pulmonary disease, and osteoporosis (Joehanes et al. Cigarette smoking has a broad impact on genome-wide and gene-specific methylation that may persist many years after smoking cessation (Joehanes et al. Epigenome-wide association studies have identified many candidate genes associated with smoking-related diseases (Lee and Pausova, 2013). In nonsmall cell lung cancer, tobacco smoke causes hypermethylation of the p16 gene promoter region associated with the duration of smoking (Kim et al. In A549 and Calu-6 lung cancer cells, exposure to tobacco smoke condensate increases tumorigenicity through the polycomb-mediated repression of Dickkopf-1 gene, which coincided with decreased H4K16Ac and increased H3K27me3 within the Dickkopf-1 promoter region (Hussain et al. Environmental chemicals that reprogram certain epigenetic events have been suggested to be at least partially involved in the pathogenesis of diseases, and the therapeutic drugs targeting distinct epigenetic modifiers have shown promising clinical outcomes in human diseases such as cancer. Many state-of-the-art technologies, including high-throughput sequencing and bioinformatics tools, have been tremendously helpful to decode the epigenome toward the ultimate goal of improving human health. Epigenetic responses following maternal dietary exposure to physiologically relevant levels of bisphenol A. Global levels of histone modifications in peripheral blood mononuclear cells of subjects with exposure to nickel. Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Occupational exposures to polycyclic aromatic hydrocarbons, and respiratory and urinary tract cancers: a quantitative review to 2005. Air pollution and cardiovascular disease: a statement for healthcare professionals from the Expert Panel on Population and Prevention Science of the American Heart Association. Mortality from obstructive lung diseases and exposure to polycyclic aromatic hydrocarbons among asphalt workers. Intellectual impairment in children with blood lead concentrations below 10 microg per deciliter. Impact of cadmium exposure during pregnancy on hepatic glucocorticoid receptor methylation and expression in rat fetus. Ubiquitination of histone H2B regulates chromatin dynamics by enhancing nucleosome stability. Urothelial carcinomas arising in arsenic-contaminated areas are associated with hypermethylation of the gene promoter of the death-associated protein kinase. Epigenetic targets of some toxicologically relevant metals: a review of the literature. International studies of prenatal exposure to polycyclic aromatic hydrocarbons and fetal growth. Lim homeobox gene, lhx8, is essential for mouse oocyte differentiation and survival. Molecular targets of epigenetic regulation and effectors of environmental influences. Epigenetic side-effects of common pharmaceuticals: a potential new field in medicine and pharmacology. Sumoylated human histone H4 prevents chromatin compaction by inhibiting long-range internucleosomal interactions. The H19/let-7 double-negative feedback loop contributes to glucose metabolism in muscle cells. Histone h3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure. Isolation and characterization of protein A24, a "histone-like" non-histone chromosomal protein. Bisphenol A interacts with the estrogen receptor alpha in a distinct manner from estradiol. Association of poly(adenosine diphosphate ribosylated) nucleosomes with transcriptionally active and inactive regions of chromatin. Influence of arsenic on global levels of histone posttranslational modifications: a review of the literature and challenges in the field. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Studies on highly metabolically active acetylation and phosphorylation of histones.
Order cialis soft 20mg overnight delivery
Effect of the infusion of glucose men's health erectile dysfunction causes discount cialis soft 20mg line, itralipid and nutramin on the initiation of rat liver regeneration after partial hepatectomy. Stem cell factor and c-kit are involved in hepatic recovery after acetaminophen-induced liver injury in mice. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Plasma hepatocyte growth factor and biliprotein levels and outcome in fulminant hepatic failure. Hepatic Defenses Against Toxicity: Liver Regeneration and Tissue Repair 393 Huh, C. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Tumour necrosis factor receptor 1 and hepatocyte regeneration in acetaminophen toxicity: a kinetic study of proliferating cell nuclear antigen and cytokine expression. The age-associated decline of glycogen synthase kinase 3beta plays a critical role in the inhibition of liver regeneration. Evidence that host size determines liver size: studies in dogs receiving orthotopic liver transplants. Vascular endothelial growth factor receptor-1 signaling promotes liver repair through restoration of liver microvasculature after acetaminophen hepatotoxicity. Role of hepatocellular regeneration in chlordecone potentiated hepatotoxicity of carbon tetrachloride. Dose- and time-dependent oval cell reaction in acetaminopheninduced murine liver injury. Expression of Notch-1 and its ligand Jagged-1 in rat liver during liver regeneration. Increased toxin-induced liver injury and fibrosis in interleukin-6-deficient mice. Differential protooncogene expression in Sprague Dawley and Fischer 344 rats during 1,2dichlorobenzene-induced hepatocellular regeneration. Specific endocrine and hormonal receptor changes associated with liver regeneration in adult rats. Liver development update: new embryo models, cell lineage control, and morphogenesis. Upregulation of calpastatin in regenerating and developing rat liver: role in resistance against hepatotoxicity. Zonal distribution of transcripts of four hepatic transcription factors in the mature rat liver. Investigation of the role of glypican 3 in liver regeneration and hepatocyte proliferation. Suppression of liver regeneration and hepatocyte proliferation in hepatocyte-targeted glypican 3 transgenic mice. In vivo and in vitro hepatotoxicity and metabolism of acetaminophen in Syrian hamsters. Immediate early detection of urokinase receptor after partial hepatectomy and its implications for initiation of liver regeneration. Presence of urokinase in serum-free primary rat hepatocyte cultures and its role in activating hepatocyte growth factor. Role of hepatocellular regeneration and hepatolobular healing in the final outcome of liver injury. Amplified interactive toxicity of chemicals at nontoxic levels: mechanistic considerations and implications to public health. Tissue repair: an important determinant of final outcome of toxicant-induced injury. Liver regeneration after partial hepatectomy: critical analysis of mechanistic dilemmas. Hepatocyte growth factor induces Wntindependent nuclear translocation of beta-catenin after Met-beta-catenin dissociation in hepatocytes. Tolerance of aged Fischer 344 rats against chlordecone-amplified carbon tetrachloride toxicity. Patterns of restoration of remnant liver volume after graft harvesting in donors for living related liver transplantation. Accelerated liver regeneration and hepatocarcinogenesis in mice overexpressing serine-45 mutant beta-catenin. Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Analysis of changes in hepatic gene expression in a murine model of tolerance to acetaminophen hepatotoxicity (autoprotection). Intact signaling by transforming growth factor beta is not required for termination of liver regeneration in mice. Amplification of chloroform hepatotoxicity and lethality by dietary chlordecone (kepone) in mice. Temporal changes in tissue repair permit survival of diet-restricted rats from an acute lethal dose of thioacetamide. Diet restriction enhances compensatory liver tissue repair and survival following administration of lethal dose of thioacetamide. Stimulated tissue repair prevents lethality in isopropanol-induced potentiation of carbon tetrachloride hepatotoxicity. Hepatic Defenses Against Toxicity: Liver Regeneration and Tissue Repair 395 Rao, M. Hepatic regeneration in peroxisome proliferator-activated receptor alpha-null mice after partial hepatectomy. Type beta transforming growth factor reversibly inhibits the early proliferative response to partial hepatectomy in the rat. Liver growth in the embryo and during liver regeneration in zebrafish requires the cell cycle regulator, uhrf1. Potentiation of carbon tetrachloride hepatotoxicity and lethality in type 2 diabetic rats. Protective effect of type 2 diabetes on acetaminophen-induced hepatotoxicity in male Swiss-Webster mice. Mechanisms of inhibited liver tissue repair in toxicant challenged type 2 diabetic rats. Hepatocyte growth factor/hepatopoietin A is expressed in fat-storing cells from rat liver but not myofibroblast-like cells derived from fat-storing cells. Alpha-fetoprotein is a predictor of outcome in acetaminophen-induced liver injury. Liver-specific loss of beta-catenin results in delayed hepatocyte proliferation after partial hepatectomy. Yes-associated protein is involved in proliferation and differentiation during postnatal liver development.
Buy generic cialis soft
Likewise erectile dysfunction cvs discount 20 mg cialis soft mastercard, b-catenin, the downstream effector of Wnt/b-catenin signaling, plays an essential role in normal hepatic growth and development. Other factors beyond activation of complex signaling cascades have been studied in the field of liver regeneration. This was associated with increased expression of remodeling molecules, such as S100 calcium-binding protein (S100A4) and miR-181b (Meng et al. Several groups have established "human liver stem cells" from normal human livers by culturing isolated hepatocytes under selective culture procedures (Herrera et al. Each of these investigations has demonstrated engraftment of human hepatic cells into mouse livers. Availability of this methodology has produced significant advances in the field of human liver stem cells. Further advances in the field of human liver regeneration have been made through the success of orthotopic liver transplants and the development of large tissue banks with liver sections from patients with end-stage liver disease. The theory that hepatocytes are involved in the "ductular metaplasia" observed in numerous human liver diseases is long-standing. Evidence for this theory in humans comes predominantly from immunohistochemical studies of liver biopsies. Since hepatocytes were the only cells to proliferate and since the hepatitis B virus shows strong tropism for hepatocytes, these authors suggested that the neoductular structures developed from infected hepatocytes. Thus, the capacity of hepatocytes to transdifferentiate into cholangiocytes appears to be enhanced when the capacity of cholangiocytes to repair injury is impaired (Michalopoulos et al. In vitro studies are beginning to reveal mechanisms relevant to hepatocyte transdifferentiation. When rat hepatocytes are cultured in roller bottles, they mass together to form hepatocyte organoid tissues (Limaye et al. Transdifferentiation also occurs when rat hepatocyte spheroid aggregates are embedded in a collagen gel matrix and treated with insulin and epidermal growth factor (Nishikawa et al. After long-term culture, the transformed hepatocytes form ductular structures with apical microvilli that are surrounded by the basement membrane, which provides in vitro evidence for the plasticity of hepatocytes. Further studies are expected to lead to a better understanding of the molecular mechanisms involved in these transformations. Cholangiocytes in normal human livers (biopsies of transplanted livers) express none of these markers. In a study of livers from pediatric patients with biliary atresia and other liver diseases, numerous cholangiocytes in bile ducts undergoing proliferation expressed multiple mesenchymal markers which were not expressed by cholangiocytes of normal pediatric livers (autopsy casesdcause of death other than liver) (Diaz et al. In this patient, biopsies of the donor liver were taken at 0, 9, 24 (days), and 9 months after transplantation. None of the markers were present in the donor liver before transplant (Robertson et al. Type I (typical) results in increased numbers of intralobular bile ducts with patent lumens that are confined to portal tract areas. Concomitantly, bile duct proliferation induces neovascularization of the hepatic artery and portal vein branches with major increases in the total volumes of the biliary tree, hepatic artery, and portal vein by 18-, 4-, and 3-fold, respectively. Alterations in vascular volumes are presumed to be due to the increased nutritional and functional demands of the enlarged biliary tree (Masyuk et al. Increased synthesis and secretion of growth factors by proliferating cholangiocytes have also been proposed to play a role in the vascular remodeling (Alvaro et al. These observations do not support the hypothesis that cholangiocyte proliferation is due to paracrine stimulation from invading leukocytes (Polimeno et al. These findings further demonstrate that secretin has autocrine and paracrine roles in the regulation of biliary growth. Concomitantly, Asbt protein expression and transport activity are elevated in large cholangiocytes, whereas de novo Asbt expression and bile salt transport occur in small cholangiocytes. In turn, cholangiocytes express basolateral P2Y and possibly P2X purinergic receptors (Dranoff et al. Comparable results occur if cholangiocytes are cultured with P2 receptor inhibitors (Jhandier et al. Continued cholangiocyte proliferation in rodents eventually leads to significant liver fibrosis and finally cirrhosis (Chang et al. Multiple studies have provided evidence that cholangiocyte proliferation is a driving force for fibrosis. Furthermore, anb6 is localized almost exclusively to cholangiocytes and "ductular" hepatocytes adjacent to fibrotic septa in end-stage liver diseases of various etiologies. Thus, anb6 integrin is proposed as a marker of fibrosis progression and a novel target for antifibrotic therapies in human liver diseases (Popov et al. Atypical proliferating ductules display similar morphology regardless of disease etiology, which suggests that this phenomenon is a general response of liver to long-standing damage. Alagille syndrome is a neonatal disease characterized by congenital intrahepatic ductopenia, and cardiac, musculoskeletal, and eye abnormalities (Piccoli and Spinner, 2001). Patients display severe jaundice and cholestasis but progression to liver cirrhosis is slow, with failure to grow the usual indication for liver transplantation (Fabris et al. Neonatal biliary atresia is due to a failure to develop major septal bile ducts or their destruction shortly after birth; biliary atresia causes severe cholestasis and rapid liver cirrhosis. Mutations in Jagged1, a ligand for Notch receptors, or in the Notch2 receptor itself, are responsible for Alagille syndrome (Fabris et al. At postnatal days 1 and 7 in the mouse, cholangiocytes forming tubular structures highly express Notch2 and Anatomy and Physiology of the Biliary Epithelium 75 adjacent portal mesenchymal cells express Jagged1 (Kodama et al. Thus, Notch signaling is essential for the formation of tubular structures during intrahepatic bile duct development. Parasympathetic innervation of the liver (evidenced by receptors for acetylcholine and vasoactive intestinal peptide) is thus considered an essential aspect of regeneration. Unraveling these complex interactions at the tissue level will provide key insights into cholangiocyte pathobiology and therapeutic options. In addition to secretin, other proteins have been shown to play significant roles in cholangiocyte proliferation and ductular reaction. Knockout of Ngn-3 reduced cholangiocyte proliferation and collagen deposition in these models (Marzioni et al. Diseases of the intrahepatic biliary tree (cholangiopathies) target the biliary epithelium and share pathogenic mechanisms such as inflammation, cholestasis, biliary proliferation, fibrosis, apoptosis, and possibly neoplastic transformation (Strazzabosco et al. Cholangiocytes from normal livers secrete several cytokines and chemokines (Komori et al. However, under disease/injury conditions, inflammatory mediators activate cholangiocytes to a reactive phenotype that participates in the pathophysiological processes by increased secretion of proinflammatory, chemotactic, and growth factors. Following activation, cholangiocyte function is modulated, antigen presentation capabilities are developed, and interactions with immune cells are enhanced (Strazzabosco et al. These and many other observations support the concept that cytokines/mediators secreted by cholangiocytes, inflammatory cells, endothelial cells, fibroblasts, and stellate cells are heavily involved in the pathogenic mechanisms of cholangiopathies (see reviews by Fava et al. Note: Discrepancies in expression levels of specific proteins between studies are indicated by more than one symbol within a column. Effects of cholangiopathies on cholangiocyte transporter expression are summarized in Table 5 (see Section Regulation of cholangiocyte function for transporters identified on cholangiocytes). Cholangiopathies have pleiotropic effects on protein expression as indicated by Table 5, where representative alterations in various types of proteins are identified. Our knowledge is very limited about whether these alterations in protein expression are primary initiators of cholangiocyte diseases or secondary effects of concomitant disease processes. These differences could explain the association observed between the duration of preservation prior to liver transplantation and the formation of bile duct strictures in liver allografts (Noack et al. The greater resistance of cholangiocytes to injury by bile salts in the intact liver can be attributed to their protection by lecithin secreted across the canaliculi of hepatocytes. Furthermore, all four of the bile salts upregulate the expression of apical Asbt, the transporter for uptake of bile salts, by a mechanism which is reversed when lecithin is added to bile salts (Tsuboi et al. These observations indicate that lecithin in bile inhibits bile salt-induced cholangiocyte injury by regulation of expression of apical and basolateral bile salt transporters, thus acting as a cytoprotective factor against accumulation of injurious bile salts (Tsuboi et al. Possible etiologies for these diseases include autoimmune diseases, genetic abnormalities, infectious agents, ischemia, and allograft rejection as well as drugs and xenobiotics, particularly their reactive metabolites (Reau and Jensen, 2008; Treinen-Moslen and Kanz, 2006).
Order generic cialis soft line
In some instances impotence exercise order cialis soft 20mg with visa, the products of biotransformation reactions (metabolites) are as active, or more active, than the original agent. Chemicals (and their metabolites) that are hydrophilic are typically excreted in urine and/or bile. Some lipophilic substances are excreted via feces in a process that involves incorporation of the lipophilic substance into micelles and subsequent biliary excretion. Hormones are responsible for coordinating the tissues of the body from conception until death (Diamanti-Kandarakis et al. As potent signaling molecules, virtually all actions of the body require hormones. Not only are these molecules responsible for reproduction but also they are essential for embryonic and fetal development, puberty, pregnancy, and aging. Cells that express these receptors are therefore responsive to the hormone, but cells that do not express the receptor are unaffected. Responses can be modulated by altering the concentration of hormones in blood or target tissues or by modulating the number of receptors. Most endogenous hormones act at exceptionally low doses, typically in nanomolar to picomolar (part-per-billion or part-per-trillion) concentrations (Vandenberg et al. The effects of hormones depend on life stage (Wallen, 2009; Heindel and Vandenberg, 2015). The same hormone, at a given dose, will have different effects on adults than it will have on developing embryos, fetuses, or neonates. The effects of hormones on adults are termed "activational" because the individual is activated to respond when exposures occur, but the effects cease when exposures are terminated. Effects during development are termed "organizational" because they can permanently alter the organizationddifferentiation, proliferation, and so ondof cells, tissues, and organs. Hormones rarely exhibit linear relationships between dose and effect over a wide range of doses (Welshons et al. This is because there is a nonlinear relationship between hormone dose and number of receptors bound, as well as a nonlinear relationship between number of receptors bound and biological effect. This principle will be discussed in more depth throughout the remainder of this article. One issue is the relationship between binding affinity and potency (Bergman et al. Binding affinity is a way to characterize the relationship between the concentration of the ligand that is required to maximally occupy the ligand-binding site of the receptor. This is quite different from the potency of a compound; potency describes the concentration of a compound that is required to produce a biological effect at a given intensity. Potency is thus endpoint specific and may not necessarily be predicted by binding affinities. For example, two compounds could have binding affinities that differ by 1000-fold; for one endpoint, they could have potencies that also differ by 1000-fold whereas for another endpoint they could be equipotent. It is often presumed that toxicants have a "threshold dose," and it is assumed (but rarely demonstrated) that no biological effects occur below this dose, whereas above the threshold, biological effects are evident. In this view, all substances are considered to be toxicants differing only in their inherent potency. Yet, the concept of thresholds in many circumstances may not apply to endocrine active substances. This is because the endocrine system is already active in living organisms that use hormones. Therefore, rather than a substance having to activate a system that is inactive (and might be considered to have some threshold for activation), the endocrine system is active and can readily be disrupted by small amounts of chemicals that activate or antagonize it. Thresholds for chemicals that act as hormone receptor agonists and antagonists are difficult, if not impossible, to prove experimentally, and it has been argued that they do not exist (Zoeller et al. Others have shown that experiments, including test guidelines used to evaluate toxicity for regulatory purposes, often lack the statistical power to appropriately evaluate whether there are biological effects below "thresholds" because as the magnitude of an observed effect decreases, the sample size required to demonstrate such an effect increases (Scholze and Kortenkamp, 2007). Hazard characterization can determine whether a compound is a reproductive toxicant, a developmental toxicant, a neurotoxicant, and so on. The number of doses that are examined will influence how well the shape of the curve is characterized. Log-linear dose responses occur when the response increases (or decreases) at a constant rate over log-increases in doses. Truly linear dose responses are rare for biological processes, although linear responses are often observed within a limited portion of the dose range tested. Sigmoidal curves are expected for many biological responses because a continued linear response at increased dose, to an infinitely high dose, is illogical; rather, a plateau in response is expected for almost all endpoints. In contrast, nonmonotonic responses are defined mathematically as ones where the sign of the slope changes over the range of doses tested (Kohn and Melnick, 2002). Nonmonotonic dose responses are sometimes mistakenly referred to as examples of hormesis. Hormesis is a descriptive word that is used to describe dose responses where one range of doses is considered to have "beneficial" effects and another range of doses induces "toxic" effects (Calabrese, 2011). In some studies, hormesis is described as "low dose stimulation, high dose inhibition. Some studies have shown that low-level radiation decreases the number of deaths compared to the background population (a low-dose "benefit"), whereas high doses of radiation increase the number of deaths compared to background (a high-dose "toxicity") (Socol et al. Dose responses that are identified as examples of hormesis are probably better described as nonmonotonic because this term provides a mathematical explanation of the shape of the dose response curve without conflating curve shape with harm or benefit (Mushak, 2007). How can nonmonotonicity be demonstrated if such mathematical descriptions of the dose response are not calculated To determine whether a dose response is consistent with hormesis, experimenters start by graphing the responses relative to the control, so responses that are increased or decreased relative to the untreated (or unexposed) group can be more easily visualized. The concept of hormesis proposes that low doses would induce responses that are often characterized as "stimulating" a "beneficial" response whereas high doses induce toxicity responses that are characterized as "harmful. Here, we demonstrate the minimum number of dose groups to calculate the slopes of two lines, as two slopes with different signs (positive or negative) must be present to draw conclusions about the presence of a nonmonotonic response. In this example, treatment dose B is the point of inflection and is used to calculate the slope of both lines. Dose responses that are consistent with nonmonotonicity have been identified using other criteria (Vandenberg and Bowler, 2014) including: 1. A significant difference is observed between the untreated control group and a mid-dose group. Similar biphasic relationships are observed for many vitamins and essential nutrients; too little or too much can induce toxicity (Querfeld and Mak, 2010). Thus, nonmonotonic dose responses are well understood in the field of nutrition science. These curves represent three criteria that can be used to determine whether a dose response is consistent with nonmonotonicity. These include: (A) a low or moderate dose is significantly different from the control, but a high dose is not. Similarly, nonmonotonicity has been observed for the relationship between hormones and a wide range of endpoints including frank diseases (Vandenberg et al.
