

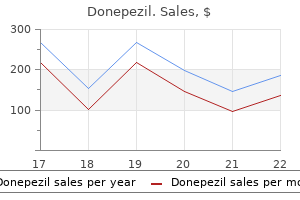
Generic donepezil 10 mg overnight delivery
The relationship between growth hormone level and sleep apnea medicine journals impact factor generic donepezil 5mg with amex, however, has remained somewhat controversial. Octreotide, a long-acting somatostatin analog, is an effective noninvasive treatment for sleep apnea in acromegaly. Sleep disturbances are common in patients with chronic renal failure with or without dialysis, particularly in those with end-stage renal disease. Fibromyalgia syndrome is characterized by diffuse muscle aches and pains not related to diseases of the joints, bones, or connective tissues; specific diagnostic criteria for this syndrome have been established. Sleep disturbances in peptic ulcer patients characteristically result from episodes of nocturnal epigastric pain. Chronic fatigue syndrome is an ill-defined, medically unexplained heterogeneous condition with multiple causes (Evengard and Klimas, 2002). The condition is characterized by insidious onset of disabling fatigue present for at least 6 months without any causes despite intense laboratory investigations. Arthralgias, myalgias, sore throat, headache, and sleep disturbances are included as minor criteria. Orthostatic hypotension and orthostatic tachycardia syndrome on a tilt-table study may be present in some of these patients (Gerrity et al. All these conditions can be associated with sleep disturbances such as insomnia, hypersomnia, and sleep-related respiratory dysrhythmia (Drouot et al. African sleeping sickness is caused by Trypanosoma gambiense or Trypanosoma rhodesiense and is transmitted to humans by the bite of tsetse flies. The patient remains somnolent in the daytime and progresses gradually into the stages of stupor and coma. Circadian disruption of plasma cortisol, prolactin, and sleep/wake rhythms is seen in the patients with the most advanced disease. The treatment of choice for patients in the meningoencephalitic stage is arsenical melarsoprol. Specific examples of psychiatric conditions that are associated with insomnia include depression (including bipolar disorder), anxiety disorders, schizophrenia, and post-traumatic stress disorder. Hypersomnia is generally present in patients with seasonal affective disorder and may also be present in some patients with depression. Many patients with eating disorders, some of whom have associated major depression, may also have significant sleep complaints. Finally, antidepressant and neuroleptic drugs used to treat psychiatric illnesses may cause sleep disruption. Assessment of sleep disturbances in psychiatric patients should follow the general guidelines discussed in this chapter. Several recent surveys found that approximately 25% of children aged 1 to 5 years have some kind of sleep problem. Behavioral insomnia of childhood includes difficulty falling asleep, staying asleep, or both, related to specific behavior such as inappropriate sleep-onset associations or inadequate limit setting. Sleep-onset association includes impairment of sleep onset because of the absence of a certain object or set of circumstances. First and foremost in the diagnosis of a sleep disorder is a detailed history, including sleep and other conditions, as outlined under Approach to a Patient with Sleep Complaints. This should be followed by a careful physical examination to uncover any underlying medical, neurological, or other causes of sleep dysfunction. Laboratory tests should include a diagnostic workup for the primary condition causing secondary sleep disturbance and a workup for the sleep disturbance itself. Various other tests are also important for assessment of a patient with sleep dysfunction (Box 102. Some young children may have obstructive hypoventilation consisting of prolonged periods of partial upper airway obstruction associated with oxygen desaturation and hypercapnia. Obstructive hypoventilation with continuous snoring as well as paradoxical breathing are commonly present in children. All apneic episodes may not be followed by cortical arousals, but they do have autonomic activation following the apneic episodes. In contrast to adults, removal of the tonsils and adenoids in children promotes symptomatic improvement. Some occurrences of sudden infant death syndrome have been linked to sleeping in the prone position, and every attempt must be made to keep the infant in the supine position. If the child or adult began to have bed wetting at least twice a week for at least 3 months after remaining dry for 6 consecutive months, then it is considered secondary sleep enuresis. Enuretic episodes occur during all stages of sleep, most commonly during the first one-third of the night. Maximum oxygen desaturation also occurs at this stage, so a daytime study cannot assess severity of symptoms. Ideally, sleep scoring should be performed manually; computerized scoring is unreliable. This manual also lists criteria for sleep and other event scoring in children older than 2 months of age. Thermistors or thermocouples are generally used to qualitatively record oronasal airflow, but these are not reliable for accurate determination of hypopnea. Most laboratories now record nasal pressure using a nasal cannulapressure transducer, which is more sensitive than a thermocouple or thermistor for detecting airflow limitation (Ayappa et al. Respiratory efforts can be recorded by use of strain gauges or inductance plethysmography. Inductance plethysmography and a piezoelectric strain gauge are the preferred methods; they can be used in a qualitative or semiquantitative fashion to monitor chest and abdominal movements. An arousal index of up to 10 is considered normal; 10 to 15 can be considered borderline. Other findings include short latency, increased time spent awake after sleep onset, and excessive snoring. Similar but less intense findings have been reported in patients with olivopontocerebellar atrophy (Chokroverty et al. Similar findings may be noted in polyneuropathies or neuromuscular junctional disorders. It also consists of four to five trials of remaining awake recurring every 2 hours. If the mean sleep latency is less than 8 minutes, it is then considered an abnormal test; values greater than this but less than 40 minutes are of uncertain significance. Underlying activity represents rhythmic ictal discharges beginning at F3-C3 (left frontocentral) and spreading rapidly to the right hemisphere, and it is accompanied by clinical seizure. Neuroimaging Studies Neuroimaging studies include anatomical and functional (physiological) studies. These studies are essential when a neurological illness is suspected of causing a sleep disturbance. A wrist-actigraphic recording from a 55-year-old healthy woman without sleep complaints. This shows a fairly regular sleep/wake schedule except one weekend night (third from top). She goes to bed between 10:30 pm and 11:00 pm and wakes up around 7:00 am except on the third day. Physiological body shifts and movements during sleep are indicated by a few black bars in the white areas. Timing of the sleep period is delayed, with sleep typically occurring from 3:00 am to about 10:00 to 11:00 am (lighter areas). A wrist actigraphic recording from a 55-year-old woman with a history of sleep-onset and maintenance insomnia. The sleep period is indicated by white areas, and wakefulness is indicated by black bars. Doppler ultrasonography is an important test for investigation of stroke due to extracranial vascular disease. In selected patients, fiberoptic endoscopy may be performed to locate the site of collapse of the upper airway, and cephalometric radiographs of the cranial base and facial bones may be obtained to assess posterior airway space or maxillomandibular deficiency. Finally, chemical control of breathing (hypercapnic and hypoxic ventilatory responses) may be needed to assess respiratory functions and control systems in patients with various neurological disorders causing dysfunction of the metabolic respiratory controllers, as well as in patients with obesity-hypoventilation (pickwickian) syndrome. A phrenic nerve and intercostal nerve conduction study may detect phrenic and intercostal neuropathy, which may cause diaphragmatic and intercostal muscle changes in some patients with neurological disorders.
Cheap donepezil 10 mg amex
Nocturnal frontal lobe epilepsy occurs exclusively during sleep at night medicine 877 generic 5 mg donepezil visa, but other types of frontal lobe seizures may be both diurnal and nocturnal. The duration is mostly less than 1 minute but sometimes may be 1 to 2 minutes with brief postictal confusion. Insomnia in epileptics may be related to frequent arousals and sleep fragmentation resulting from nocturnal seizures and interictal epileptiform discharges, depression, anxiety, associated primary sleep disorders, and a specific effect of some antiepileptic medications. There is probably an increased prevalence of sleep apnea in epilepsy, as reported by scattered case reports, but systematic study to determine the true preva- lence of sleep apnea has not been undertaken (Chokroverty et al. Degenerative Dementia and Sleep Dysfunction Dementia is characterized by progressive deterioration of memory and cognition followed by language dysfunction, hallucinations, other psychotic features, depression, and sleep disturbance. Sleep dysfunction with or without abnormal motor activity during sleep is increasingly recognized in patients with irreversible chronic dementing illnesses (Chokroverty, 2009a; Chokroverty et al. The major sleep disturbances in dementing illnesses include insomnia, hypersomnia, circadian sleep/wake rhythm disorders, excessive nocturnal motor activity, "sundowning," and respiratory dysrhythmias. Synucleinopathies are a group of disorders with abnormal deposition of -synuclein in the cytoplasm of neurons or glial cells, as well as in extracellular deposits of amyloid. In addition to sundowning, these patients often sleep early in the evening, waking up frequently and staying awake most of the night. Sleep-onset and maintenance insomnia, and sleep fragmentation are common (Peeraully et al. Most commonly, sleep disruption with fragmentation is noted with increasing disease severity. Fatal familial insomnia is a rare and rapidly progressive autosomal dominant prion disease with a missense mutation at codon 178 of the prion protein gene (PrP) (Montagna, 2011). Clinical manifestations are impaired control of the sleep/wake cycle, including circadian rhythms; autonomic and neuroendocrine dysfunction; and somatic neurological, cognitive, and behavioral manifestations. Profound sleep disturbances and, in particular, severe insomnia are noted from the very beginning of the illness. This abnormal sleep pattern is associated with dream-enacting behavior in the form of complex gestures and motions and myoclonus. Autonomic function tests reveal evidence of sympathetic hyperactivity with preserved parasympathetic activity. Persistently elevated serum catecholamine levels associated with abnormal secretory patterns of growth hormone, prolactin, and melatonin are noted. Plasma melatonin levels progressively decrease, and in the most advanced stage of the illness, there is a complete abolition of melatonin rhythm. Somatic neurological manifestations are present in all cases, particularly in the later stage of the illness and consist of dysarthria, dysphagia, ataxia, evidence of pyramidal tract dysfunction, myoclonus, tremor, and bizarre astasia-abasia (Cortelli et al. There are no spongiform changes, except in those with the longest duration of symptoms, who show mild to moderate spongiform degeneration of the cerebral cortex. In Creutzfeldt-Jakob disease, prominent sleep disruption is noted even in the early stage and is characterized by sleep-onset and maintenance insomnia with daytime hypersomnolence. Sleep Disorders Associated with Neuromuscular Disorders Clinicians first became aware of sleep-related respiratory dysrhythmias in patients with neuromuscular diseases by observing hypoventilation in poliomyelitis. Sleep disturbances in neuromuscular diseases are generally due to respiratory dysrhythmias associated with these diseases (Bhat et al. In neuromuscular disorders, sleep disturbances are due to involvement of respiratory muscles, phrenic and intercostal nerves, or neuromuscular junctions of the respiratory and oropharyngeal muscles. In addition to the sleep-related respiratory dysrhythmias, some patients- particularly those with painful polyneuropathies, muscle pain, muscle cramps, and immobility due to muscle weakness- may complain of insomnia. Patients with neuromuscular diseases often complain of breathlessness, particularly in the supine position. Respiratory disturbances are generally noted in the advanced stage of primary muscle disorders or myopathies, but respiratory failure may appear in an early stage. They may also occur in other congenital or acquired myopathies, mitochondrial encephalomyopathy, and polymyositis. In polyneuropathies, involvement of the nerves supplying the diaphragm and the intercostal and accessory muscles of respiration may cause breathlessness on exertion and other respiratory dysrhythmias. These may worsen during sleep, causing sleep fragmentation and daytime hypersomnolence. Patients with myasthenia gravis may have central, obstructive, and mixed apneas and hypopneas accompanied by oxygen desaturation (Chokroverty, 2011a). A sensation of breathlessness on awakening in the middle of the night and early-morning hours may indicate respiratory dysfunction. Sleep-related hypoventilation and sleep apnea in neuromuscular junctional disorders may be severe enough to require assisted ventilation (Gonzalez et al. Sleep and Spinal Cord Diseases Sleep disturbances related to respiratory dysfunction can occur in some patients with high cervical spinal cord lesions. The most common symptom is hypersomnia due to sleeprelated respiratory arrhythmias. Occasionally, patients with spinal cord diseases may complain of insomnia as a result of immobility, spasticity associated with flexor spasms, neck pain, and central pain syndrome. Respiratory disturbances worsening during sleep may occur in many patients during the acute and convalescent stages of poliomyelitis. Some are left with the sequelae of sleep-related apnea or hypoventilation, requiring ventilatory support, especially at night. Another group of patients develops symptoms decades later that constitute postpolio syndrome, in which sleep disturbances and sleep apnea or hypoventilation have also been noted (Bhat et al. Postpolio syndrome is manifested clinically by increasing weakness or wasting of the previously affected muscles and by involvement of previously unaffected regions of the body, fatigue, aches and pains, and sometimes symptoms secondary to sleep-related hypoventilation. It is characterized by progressive degeneration of both upper and lower motor neurons, manifesting as a varying combination of lower motor neuron. Insomnia may be present in some patients, which is related to other factors such as decreased mobility, muscle cramps, anxiety, and difficulty swallowing. There is no significant relationship between bulbar involvement and severity of sleep-disordered breathing or other types of respiratory events. In addition, degeneration of central respiratory neurons may occur, causing central and upper airway obstructive sleep apneas. Sleep apnea, snoring, and stroke are intimately related; sleep apnea may predispose to stroke, and stroke may predispose to sleep apnea. There is an increased frequency of sleep apnea in both infratentorial and supratentorial strokes (Bassetti and Hermann, 2011; Bassetti and Valko, 2006; Davis et al. Many confounding variables such as hypertension, cardiac disease, age, body mass index, smoking, and alcohol intake, which are common risk factors for sleep apnea and stroke, must be considered before a clear relationship can be established among these conditions. Most sleeprelated headaches are daytime headaches that may also occur during sleep. An uncommon type of headache syndrome called hypnic headache syndrome is described in patients older than 50. The headache awakens the patient from sleep, lasts for at least 15 minutes with a range up to 190 minutes, and with a frequency of at least 15 times per month. However, no studies have adequately addressed the sleep/wake abnormalities in these patients after recovery from coma or in patients after minor brain injuries that did not result in coma. Many of these patients experience the so-called postconcussion syndrome, which is characterized by a variety of behavioral disturbances, headache, and sleep/wake abnormalities. A few reports list subjective complaints of sleep disturbance but do not include formal sleep studies. Sleep Disorders in Autonomic Failure: Multisystem Atrophy (Shy-Drager Syndrome) In a consensus statement by the American Autonomic Society and the American Academy of Neurology (Gilman et al. Striatonigral degeneration is the name used when the predominant feature is parkinsonism, whereas olivopontocerebellar atrophy is used when the cerebellar features are the predominant manifestations. The term Shy-Drager syndrome is still used when the autonomic feature is predominant. The current diagnostic criteria including those derived from the second consensus statement (Gilman et al. This finding may lead to a future consensus statement to revise the diagnostic criteria.
Discount generic donepezil uk
However treatment interventions purchase genuine donepezil, the process usually involves all layers DisordersofBones,Joints,Ligaments,andMeninges 1763. Some argue that the eponym Mollaret meningitis should be reserved for those cases without an identified causative organism. Idiopathic hypertrophic pachymeningitis refers to those cases for which cultures, serology, and dural biopsy fail to reveal a causative infection, tumor, or systemic inflammatory disease. These patients can present with headache, cranial neuropathies, ataxia, or seizures. Dural biopsy shows small mature lymphocytes, plasma cells, and epithelioid histiocytes. Patients can improve after treatment with corticosteroids, sometimes supplemented with drugs such as azathioprine and methotrexate. The clinical diagnosis is difficult, but the neuroradiological abnormalities are striking. These paramagnetic signal changes represent encrustation of the brain surfaces with hemosiderin. The adjacent neural tissue atrophies, with accumulation of ferritin in microglia and Bergmann cells in the cerebellum. Superficial siderosis is secondary to chronic or recurrent blood leakage into the subarachnoid space. Some patients with spinal dural leaks develop both superficial siderosis and intracranial hypotension (Kumar et al. Treatment relies on identifying and arresting the source of bleeding; chelation therapy does not appear to be effective. UveomeningitisSyndromes the combination of chronic or recurrent meningitis and uveitis has a specific differential diagnosis (Yeh et al. Often, ophthalmological characterization of the uveitis can refine the differential diagnosis. For example, the uveitis of Vogt-Koyanagi-Harada syndrome is bilateral and often causes retinal elevations and retinal pigmentary changes. Vogt-Koyanagi-Harada syndrome also causes skin and hair findings such as vitiligo, poliosis, and focal alopecia. The American College of Rheumatology classification criteria for the diagnosis define pain as widespread when it is bilateral, above and below the waist, and axial. Typically, patients have multiple symptoms including fatigue, stiffness, nonrestorative sleep, headaches, and mood disorders (Bennett, 2009). Patients may have many symptoms of neurological import such as weakness, paresthesia, imbalance, and dizziness and often have cognitive complaints regarding concentration, memory, and multitasking. SuperficialHemosiderosis Superficial hemosiderosis is a rare disorder that causes slowly progressive cerebellar ataxia, mainly of gait, and sensorineural deafness, sometimes accompanied by manifestations of myelopathy such as spasticity, brisk reflexes, extensor plantar responses, bladder disturbance, or sensory signs (Kumar et al. The fibromyalgia syndrome: myofascial pain and the chronic fatigue syndrome, in: Kelley, W. Diagnostic neurological investigations such as brain imaging, muscle biopsy, or electrodiagnostic studies are normal or show minor nonspecific abnormalities. Behavioral and biological factors both contribute to the clinical presentation of the syndrome. Neuroscientific research on the pathogenesis of fibromyalgia has examined muscle, sleep, neuroendocrine function, and central pain processing, including studies using functional brain imaging (Nebel and Gracely, 2009). Symptoms and signs of fibromyalgia can occur in association with autoimmune diseases such as systemic lupus erythematosus or other systemic illness such as hypothyroidism. The pathogenic role of trauma, on-the-job injury, or workplace stress is controversial. Small short-term controlled studies have suggested that some patients benefit from acupuncture. Ventral fusion versus dorsal fusion determining the optimal treatemt for cervical spondylotic myelopathy. Assessment: use of epidural steroid injections to treat radicular lumbosacral pain: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Effects of surgery on the sensory deficits of syringomyelia and predictors of outcome: a long-term prospective study. Osteogenesis imperfecta: recent findings shed new light on this once well-understood condition. A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. Rotational vertebral artery insufficiency resulting from cervical spondylosis: case report and review of the literature. A review of the cases presenting in a regional spine injuries unit in the north east of England over a 5-year period. Surgery for low back pain: a review of the evidence for an American Pain Society Clinical Practice Guideline. Indications for magnetic resonance imaging in presumed adolescent idiopathic scoliosis. Clinical outcomes after acute osteoporotic vertebral fractures: a 2-year non-randomised trial comparing percutaneous vertebroplasty with conservative therapy. Performance of various criteria sets in patients with inflammatory back pain of short duration; the Maastricht early spondyloarthritis clinic. Position statement on percutaneous vertebral augmentation: a consensus statement developed by the American Society of Interventional and Therapeutic Neuroradiology, Society of Interventional Radiology, American Association of Neurological Surgeons/Congress of Neurological Surgeons, and American Society of Spine Radiology. Asymptomatic Chiari type I malformations identified on magnetic resonance imaging. Specific entities affecting the craniocervical region: osteogenesis imperfecta and related osteochondrodysplasias: medical and surgical management of basilar impression. Tethered cord syndrome in childhood: diagnostic features and relationship to congenital abnormalities. Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and Chiari malformation type I in patients with hereditary disorders of connective tissue. Epidemiology, etiology, pathogenesis, and diagnosis of recurrent bacterial meningitis. Neurovascular complications of Marfan syndrome: a retrospective, hospital-based study. Chiari I malformation redefined: clinical and radiologic findings for 364 symptomatic patients. Investigations into the association between cervicomedullary neuroschisis and mirror movements in patients with Klippel-Feil syndrome. In these complicated clinical settings, clinicians may often turn to serological, radiological, and elec trodiagnostic studies to aid in diagnosis. The sections that follow cover some anatomical features relevant to an understanding of the pathological conditions that affect the nerve roots, as well as specific nerve root disorders. Anatomical Features Each nerve root is attached to the spinal cord by four to eight rootlets that are splayed out in a longitudinal direction (Rankine, 2004). The dorsal rootlets are attached to the spinal cord at a welldefined posterolateral sulcus, whereas the ventral rootlets are more widely separated and emerge over a greater area of the anterior surface of the spinal cord. The dorsal ramus innervates the deep posterior muscles of the neck and trunk (the paraspinal muscles) and the skin overlying these areas. The ventral ramus, depending on its spinal segment, contributes to an intercostal nerve, or to the cervical, brachial, or lumbosacral plexi and thereby sup plies the trunk or limb muscles. Directly adjacent to the spinal cord, the nerve roots lie freely in the subarachnoid space, covered by a thin root sheath, composed of a layer of flattened cells, that is continuous with the pial and arachnoidal coverings of the spinal cord. Com pared with spinal nerves, the roots have many fewer connec tive tissue cells in the endoneurium and considerably less collagen. Moreover, they lack an epineurium, as this dense connective tissue layer is contiguous with the dura mater. A capillary network derived from the radicular arteries provides the blood supply to the spinal nerve roots (Becske and Nelson, 2009). Where the nerve roots form the mixed spinal nerve, the pial covering of the root becomes continuous with spinal nerve perineurium, and the nerve takes the dural nerve root sheath through the intervertebral foramen to become continuous with the epineurium of the mixed nerve.
Order donepezil from india
Objective findings frequently elude sophisticated vestibular testing acute treatment discount donepezil american express, which only occasionally reveals nystagmus of possible central or peripheral etiology. Classic downbeat nystagmus is rarely seen even with tonsillar herniation as striking as 20 mm. Headache symptoms sometimes respond to carbonic anhydrase inhibitors, simple approaches to associated cervical muscle spasms, or head elevation during sleep. Minor traumatic events can aggravate symptoms, and patients should avoid activities that risk head and neck injury. For women with syringomyelia, a cesarean section is advisable to avoid the intrathoracic and abdominal pressure elevations associated with vaginal delivery. Surgical decompression of the posterior fossa is appropriate treatment for the minority of patients who have significant functional impairment due to headache despite medical therapy or have progressive syringomyelia. It is not indicated for symptoms limited to chronic fatigue, musculoskeletal pain, or vertigo, or for prophylaxis against worsening of headache or of syringomyelia. Kula (2006) discusses treatment in detail and provides a comprehensive management algorithm. The most extreme form is anencephaly, characterized by absence of the entire cranium at birth; the undeveloped brain lies at the base of the skull as a small vascular mass without recognizable nervous structures. SpinaBifidaOcculta Spina bifida occulta is the most common and least symptomatic (usually asymptomatic) form of dysraphism. In this anomaly, the vertebral elements fail to fuse posteriorly, but the thecal and neural elements remain within the spinal canal. Orthopedic foot deformities, urinary or rectal sphincter dysfunction, or focal neurological abnormalities can indicate that the spina bifida occulta is associated with compression or malformation of neural tissues or with spinal cord tethering. At times the skin and vertebral canal are open, and a sac of meninges is directly visible. If the sac contains nerve roots or spinal cord, it is a myelomeningocele; if neural elements are absent from the sac, it is a meningocele. Neurological deficits are directly related to the anatomical extent of the malformation and vary from insignificant to grave. The extent of brainstem herniation is variable, including portions of the medulla or even of the pons. Hydrocephalus and syringomyelia are common accompanying features, and patients often have various associated anomalies such as a small posterior fossa, kink in the medulla, and polymicrogyria. These infants are at risk for later development of tethered cord syndrome, spinal dermoid, and epidermoid inclusion cysts. An important cause is maternal folate deficiency, and most cases would be prevented if women with childbearing potential routinely took folic acid daily. Other risk factors include family history of neural closure defects and maternal treatment with some antiepileptic drugs such as valproic acid. Planning treatment for affected infants, potentially including surgery, is difficult. Initial surgical treatment in utero or in the neonatal period can provide cosmetic repair and decrease the risk for meningitis. Any existing myelopathic or radiculopathic neurological deficit is likely to persist after surgery. Some patients, especially infants with progressive brainstem dysfunction, are treated with decompression of the rostral spinal canal. Less than 30% of such patients survive beyond the first year, and long-term problems including mental retardation and paraplegia are often severe. Few patients with myelomeningocele are mentally normal, but most of those with lumbar meningocele are. TetheredCordSyndromes Congenital abnormalities of the spinal cord or cauda equina can result in spinal cord tethering, in which stretching and tension develops within the cord tissue as the spinal column elongates during early life, resulting in the conus medullaris being found at an abnormally low vertebral level (Michelson and Ashwal, 2004) (Box 105. A child or even an adult with these abnormalities can develop progressive neurological dysfunction due to traction on the cord or nerve roots. One presentation is lower motor neuron dysfunction in one or both legs, but patients can also have sensory loss, upper motor neuron signs, orthopedic foot deformities, and scoliosis. A tethered spinal cord can also cause isolated sphincter dysfunction as subtle as intermittent urinary incontinence. The so-called occult tethered cord syndrome is an area of controversy (Drake, 2006; Selden, 2006). A few cases of cerebellar tonsillar herniation seem to be due to occult cord tethering; other features are syrinx development below the T5 level and scoliosis (Milhorat et al. Supporting the role of cord tethering as a cause of the tonsillar descent are reports of increasing herniation of the cerebellar tonsils with somatic growth, cerebellar prolapse following Chiari decompression surgery, and anatomical improvements including ascent of the conus medullaris, ascent of the cerebellar tonsils, and resolution of brainstem elongation following section of the filum terminale. Dandy-WalkerSyndrome Dandy-Walker syndrome results from the failure of development of the midline portion of the cerebellum. A cyst-like structure associated with a greatly dilated fourth ventricle, expanding the midline is often seen. The malformation typically causes the occipital bone to bulge posteriorly and displaces the tentorium and torcula upward. The cerebellar vermis is aplastic, and the corpus callosum may be deficient or absent. There is usually dilation of the aqueduct as well as the third and lateral ventricles. In most instances, the division is located in the lower thoracic or lumbar regions. Diastematomyelia is often accompanied by skin abnormalities such as a tuft of hair at the level of the lesion. Neurological deficits, scoliosis, and congenital foot deformities are more common in type I. Bony and cartilaginous spurs between the hemicords are also more common with type I. Finally, surgical repair is more effective in type I and can be combined with distal untethering if a tethered cord is present as well. The spur tethers the spinal cord, leading to progressive neurological dysfunction when the vertebral column lengthens during growth. The diagnosis can often be suspected on plain radiography, which shows widening of the interpedicular distance and a posterior bony bridge at the level of the lesion. Surgical therapy consists of attempts to free all structures tethering the cord by removing the spurs and dura in the cleft and cutting the filum terminale if abnormally tethered. When fluid dissects into the surrounding white matter forming a cystic cavity or syrinx, the term syringomyelia is applied. Hydromyelia and syringomyelia often coexist, and many physicians use the terms interchangeably. The central canal of the spinal cord is normally widely open during embryonic life and becomes atretic after birth. ClinicalPresentation the prototypical presentation of a symptomatic syrinx is the combination of lower motor neuron signs at the level of the lesion (usually in the arms or lower cranial nerves), a dissociated suspended sensory loss (impaired pain and temperature sensation but preserved light touch, vibration, and position sense in a cape or hemicape distribution on the arms and upper trunk), and spinal long-tract dysfunction below the level of the lesion. However, few patients show this total picture, and the clinical features vary with the size, location, and shape of the cavity; the rapidity of its evolution; and any associated neurological conditions. Symptoms are more related to the pace of evolution of the syrinx than to its absolute size. Otherwise healthy patients with slitlike syrinx cavities may present with severe localized spinal and radicular pain. Other patients with syrinx cavities displacing as much as 90% of the spinal cord mass may be virtually asymptomatic. Common complaints include neck ache, headache, back pain, radicular pain, and areas of segmental dysesthesia. Painful dysesthesias are most likely to occur at or adjacent to the caudal extent of the syrinx cavity. Some patients have trophic changes corresponding to segmental loss of pain sensation. Syringomyelia can cause neuropathic monoarthritis (Charcot joint), most commonly in a shoulder or elbow. When the syrinx enlarges as an asymmetrical localized paracentral outpouching from the hydromyelia, particularly at its cranial or caudal ends or at its level of greatest axial cross-section, the paracentral extensions often lead to local segmental signs such as cranial nerve dysfunction in patients with syringobulbia, as well as segmental lower motor neuron signs and dissociated sensory changes at the level of spinal involvement.
Buy donepezil overnight delivery
For example treatment quotes and sayings best buy donepezil, a person who is able to speak in full sentences and engages in communication but whose to-and-fro conversation with others fails, and whose attempts to make friends are odd and typically unsuccessful. Restricted,repetitivebehaviors 1303 90 Inflexibility of behavior, extreme difficulty coping with change, or other restricted/repetitive behaviors markedly interfere with functioning in all spheres. Level 2 "Requiring substantial support" Inflexibility of behavior, difficulty coping with change, or other restricted/repetitive behaviors appear frequently enough to be obvious to the casual observer and interfere with functioning in a variety of contexts. Level 1 "Requiring support" Inflexibility of behavior causes significant interference with functioning in one or more contexts. Some children fail to process language and, despite normal hearing, appear deaf (verbal auditory agnosia) (Deonna et al. Higher-functioning children sometimes talk too much-they talk to talk (semantic and pragmatic deficits) but do not use language to communicate (pragmatics). Prosody is frequently impaired, as evidenced by mechanical, excessively rapid, monotonic, high-pitched, and/ or poorly modulated speech. Those with conversational language do significantly better than children with little or no language. Nonverbal cognitive ability at age 2 was generally the strongest predictor of age 5 language, while, at age 3, communication scores were a stronger predictor of age 5 language for children with autism. Play ranges from repetitive stereotypic and indiscriminate sensory use of objects (mouthing, rubbing, etc. Pretend play is often rudimentary, even in higher-functioning young children, involving, for example, simple role taking (Waterhouse et al. Such children do not follow their parents around, run to greet them, or seek comfort. These children tend to be of low intelligence, have poor verbal and nonverbal communication skills, and exhibit little symbolic play. These children do not make social approaches, but they accept them when made by others. They engage in some pretend play and join in games, but they take a passive role. Children who are interactive but odd make spontaneous social approaches to others, but in a peculiar manner. Pragmatic language skills are impaired; for example, questions are conversation openers. Many persons on the autistic spectrum are relatively unaware of their social ineptitude except to the extent others tease them. A restricted range of behaviors, interests, and activities is another hallmark feature of autism. In lower-functioning children, these tend to consist of repetitive stereotyped behaviors like twirling, rocking, flapping, licking, and opening and closing doors. By age 2 years, it is expected that a qualified professional can reliably make the diagnosis. The American Academy of Pediatrics recommends screening all children at well-child visits at 18 months and at 24 months of age (Johnson et al. A recent toddler module was designed to assess children who are 18 months of age upwards. Macrocephaly occurs in about a third of children with autism and generally becomes apparent around the age of 1 to 3 years. Skin examination requires careful attention, given high co-occurrence with tuberous sclerosis (Gillberg, 2010). Other conditions include purine and pyrimidine abnormalities, Smith-Lemli-Opitz syndrome, and lysosomal storage disorders. The extent of the evaluation for an underlying metabolic disorder depends on clinical suspicions and the relevance to family counseling. Double syndromes: autism associated with genetic, medical and metabolic disorders. It is important to consider medical causes for any change in behavior, especially in those individuals who are nonverbal or with limited language capability. Examples of such medical conditions include, but are not limited to , the following: pain (due to migraine headaches, ear infection, fractures, etc. Epilepsy the association of epilepsy with autism provided one of the first clues to suggest that autism was a neurodevelopmental disorder of brain function. The significance of these abnormalities, especially in the absence of clinical seizures, is quite unclear. At the present time, there are no data to support the use of antiepileptic drugs or epilepsy surgery in the treatment of these abnormalities in the absence of clinical seizures (Tuchman, 2004). Having an etiologic diagnosis offers clinicians the opportunity to provide better guidance regarding recurrence risks, as well as prognostic information and obviates the need for additional testing. Furthermore, a specific diagnosis can alert clinicians to be aware of other comorbidities associated with certain genetic syndromes. However, knockout mouse models of these mutations do not show the full range of autistic symptoms. Based on the core symptoms of autism, neuropathological abnormalities would be anticipated and are found in regions important to social function (frontal lobe, superior temporal cortex, parietal cortex and amygdala), language function (language cortex), and repetitive behaviors and stereotypies (orbital frontal cortex and caudate) (Amaral et al. Functional imaging studies demonstrate that neural systems related to social functioning, such as emotional face recognition, are abnormal (Corbett et al. Abnormalities of mirror neurons are also seen when subjects imitate and observe emotions (Rizzolatti and Fabbri-Destro, 2010). Studies of brain structure have implicated multiple events in the prenatal and postnatal brain development, particularly neuronal organizational events. Courchesne and colleagues found focal patches of abnormal laminar cytoarchitecture and cortical disorganization of neurons, but not glia, in the prefrontal and temporal cortical tissue from 10 of 11 children with autism and from one of 11 unaffected children, supporting a probable dysregulation of layer formation and layer-specific neuronal differentiation prenatally (Stoner et al. Increased total brain volume, primarily due to increased white matter, is the most frequently replicated imaging finding (Verhoeven et al. In contrast to other white-matter structures, both volume and density of the corpus callosum are reduced (Hardan et al. Imaging studies also highlight the dissociation between whitematter tract overgrowth and gray-matter dendritic and synaptic underdevelopment. Spectroscopy studies suggest that the gray matter is abnormal and dendritic arborization and synaptosome density reduced. Some investigators speculate that graymatter abnormalities trigger the white-matter overgrowth (Williams and Minshew, 2007). At the cytoarchitectonic level, minicolumns that determine connectivity are abnormal, especially in the dorsolateral prefrontal cortex (Casanova et al. The hyperconnected local networks may become partially isolated and acquire novel functional properties. By contrast, the decrease in long-range connections could explain the problems with top-down control and integration (Williams and Casanova, 2010). A variety of other interventions are used to target specific areas of development. Psychiatric comorbidities commonly develop: depression, bipolar disorder, anxiety, schizophrenia, and even catatonia (Billstedt et al. A minority of patients, usually the higher-functioning group, improve significantly during adolescence. Approximately two-thirds of adults with autism show poor social adjustment (limited independence in social relations), and half require institutionalization. Even though higher-functioning individuals with autism (including those previously diagnosed as Asperger syndrome) had the best outcome, only 15% to 30% had fair to good outcomes, and only 5% to 15% became competitively employed, led independent lives, married, and raised families. Psychopharmacologic interventions for repetitive behaviors in autism spectrum disorders. Specifically, the diagnosis requires that the following criteria are met: (1) Deficits in intellectual functioning. Clinical interview with the individual and a collateral contact who knows the individual well can help assess adaptive functioning, as can standardized measures of adaptive behaviors. Auditory and visual function must be determined, since these are common comorbidities. If a child was born in a locale without universal newborn screening, consider a screening metabolic evaluation that includes a capillary blood gas, serum lactate and ammonia levels, serum amino acids and urine organic acids, and thyroid function studies.

Alpinia. Donepezil.
- Are there any interactions with medications?
- How does Alpinia work?
- Dosing considerations for Alpinia.
- What is Alpinia?
- Intestinal gas, infections, spasms, fever, reducing swelling (inflammation), and other conditions.
- Are there safety concerns?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96299
Buy generic donepezil pills
Medical therapy can be administered prophylactically to prevent attacks of migraine or symptomatically to relieve the pain medications that cause weight loss 10 mg donepezil otc, nausea, and vomiting of an attack. Other indications for prophylaxis include severe or prolonged neurological symptoms or lack of response to symptomatic treatment. In general, prophylaxis should be considered if attacks occur as often as 1 to 2 days a week. Administration of sumatriptan can be oral, intranasal, and by subcutaneous injection (Tables 103. For patients who had no significant pain relief after 1 hour, administration of a second dose of 6 mg provided little further benefit. Side effects of sumatriptan by injection include local reaction at the injection site, usually of mild or moderate severity, and a transient tingling or flushed sensation that may localize or generalize. A more unpleasant sense of heaviness or pressure in the neck or chest occurs in a small percentage of recipients. However, because sumatriptan has been shown to produce a minor reduction in coronary artery diameter, it should be used with caution in patients who have significant risk factors for coronary artery disease and should not be given to patients with any history suggestive of coronary insufficiency. It is also contraindicated in patients with untreated hypertension, ischemic or vaso-occlusive cerebrovascular disease, peripheral vascular disease and in those using ergot preparations. It is contraindicated in women during pregnancy and in patients with hemiplegic migraine or migraine with brainstem aura (previously "basilar-type migraine"). Per the American Academy of Pediatrics, at present, sumatriptan belongs to the group of medications usually compatible with breast feeding. The potential side effects are quite similar: tingling, flushing, and a feeling of fullness in the head, neck, or chest. They are not safe when administered within 24 hours of ergot preparations or other members of the triptan class. At this time, no evidence exists to allow accurate prediction of which of these agents will be most effective in a given patient. A few practical guidelines are available, based on the clinical situation and knowledge about available agents. If severe nausea or vomiting occurs early in an attack, the parenteral or intranasal routes should be used. Some patients may prefer nasal or injectable routes (sumatriptan and Symptomatic Treatment Start symptomatic treatment as early in the development of an attack as possible. If an aura is recognized, patients should take medications during it rather than waiting for the pain to begin. It must be recalled, though, that once the attack is fully developed, oral preparations are almost always less effective because of decreased gastrointestinal motility and poor absorption. If vomiting develops, alternative routes of administration are often necessary (suppository, intranasal, and/or injections). For many patients, a simple oral analgesic such as aspirin, acetaminophen, naproxen, ibuprofen, or an analgesic combination with caffeine may be effective. Caffeine aids absorption, helps induce vasoconstriction, and may reduce the firing of serotonergic brainstem neurons. However, the use of caffeine-containing combination analgesics more than 2 days per week may lead to increased incidence of headaches. For patients with benign but intolerable side effects from this group of medications, consider naratriptan, almotriptan, or frovatriptan, given their favorable side-effect profiles. If one agent fails, it seems reasonable, barring major side effects, to try another agent in the class. Since there is evidence that some of these agents have a lower oral bioavailability when taken by patients with migraine, both during an attack and interictally (Aurora et al. Although increasingly less available and supplanted in many cases by newer agents, ergot preparations still have a role in the symptomatic treatment of migraine. Oral preparations are far less effective than those given rectally or parenterally. If selected for use, 2 mg of ergotamine tartrate by mouth should be taken as soon as the patient recognizes the symptoms of an acute migraine attack. This dose, combined with a simple oral analgesic-caffeine combination, can be taken again in 1 hour. Possibly a better regimen, but inconvenient and unpleasant to some patients, is ergotamine tartrate by rectal suppository. The patient should be instructed to insert a 1- or 2-mg rectal suppository of ergotamine tartrate at the onset of the aura or pain and take a simple analgesic orally. Experience over the course of several attacks is useful to determine the amount of ergotamine needed to obtain relief. If nausea is troublesome, metoclopramide in doses of 10 mg orally aids absorption of the ergotamine tartrate and may prevent vomiting. Analgesics in rectal suppository form include aspirin and acetaminophen, either of which may provide some relief. With frequent attacks of migraine, care must be taken to avoid the vicious cycle of medication overuse headache (discussed elsewhere in this chapter). If more than 6 mg of ergotamine is required per week, use an alternative preparation. Ergotamine must be used cautiously by patients with hypertension and those with peripheral vascular disease. It is contraindicated in patients with coronary artery disease and in women who are pregnant. It is unwise to administer ergotamine to patients in whom the aura is particularly prolonged or characterized by a major neurological deficit. The fear of potentiating the vasospasm to the point of cerebral infarction may be unjustified, but avoid the potential risk by withholding potent vasoconstrictors. As an alternative to ergotamine in the symptomatic treatment of migraine, the sympathomimetic agent, isometheptene mucate, is useful. It is available in proprietary preparations combined with acetaminophen and dichloralphenazone and has the advantages of not increasing nausea and being well-tolerated, but it may fail to give relief for severe attacks. Its poor oral bioavailability limits its administration to the parenteral and intranasal routes (see Tables 103. This medication should be considered when nausea and vomiting limit the use of oral medications or when other medications are ineffective. Increased nausea in some patients may require combination with an antiemetic agent. Symptomatic treatment of migraine with typical aura is essentially the same as that described previously, although some data suggests subcutaneous sumatriptan may not be effective if taken during the aura phase before headache onset. While there are no concrete guidelines currently available for status migrainosus treatment, several strategies have been used by some in the past. Sumatriptan, 6 mg subcutaneously, may provide relief of both the headache and the associated symptoms. Evidence for the efficacy of magnesium sulfate as an acute treatment for migraine generally favors only individuals with aura, where infusion of 1 g of magnesium sulfate may result in relief. The dose may be repeated twice at 15-30 minute intervals but the maximum dose should not exceed 37. The latter agent often produces hypotension, and patients should first receive a bolus of 250 to 500 mL of 5% dextrose in one-half normal saline. Some patients develop acute extrapyramidal symptoms after treatment with neuroleptic agents. The neuroleptic agents do produce sedation, and patients should be advised not to operate a motor vehicle after treatment. Injectable ketorolac, 60 mg given intramuscularly, is another alternative to the narcotic or sedative agents. A single dose of dexamethasone combined with other parenteral antimigraine agents is useful for the emergency room treatment of attacks of intractable migraine. Intravenous infusion of valproate sodium 500 mg in normal saline over 1 hour is yet another potentially efficacious abortive option. In a female patient, obtaining a pregnancy test prior to administering this treatment must be considered due to the potential for teratogenic effects. When status migrainosus occurs, dehydration, tiredness due to lack of sleep, and continued pain may necessitate admission to a hospital to terminate the attack.
Buy generic donepezil pills
Postencephalitic Parkinsonism Between 1916 and 1927 symptoms of depression order donepezil 10mg otc, a worldwide epidemic of encephalitis lethargica killed approximately 250,000 persons and left an additional 250,000 with chronic disability. These survivors of the acute illness developed parkinsonism, usually within 10 years of the infection. Physiological tremor appears to originate in the heartbeat, mechanical properties of the limbs, firing of motoneurons, and synchronization of spindle feedback. It is usually not noticeable except with elec trophysiological recording, but its amplitude is accentuated by fatigue, anxiety, fear, excitement, stimulant use, and medical conditions such as hyperthyroidism (Box 96. In populationbased studies, the prevalence increases steadily with age, occurring in up to 10% of patients older than 60 years of age. The median age at onset is 15 years, but there is a bimodal distribution, and virtually all patients are symptomatic by age 65. The typical patient becomes aware of a barely perceptible postural or action tremor, usually in the distal arms and hands. Head tremor (titubation) is milder than limb tremor and is predominantly of a sidetoside, "nono" type. The kinetic tremor is higher in amplitude than the postural tremor and is the major determinant of disability. A striking improvement after ingestion of a small amount of ethanol is seen in 50% of patients and may be helpful in diagnosis (Mostile and Jankovic, 2010). Only a frac tion of affected persons seek medical attention, and there is often a long latency from onset to presentation for care. This results from decreased attenuation of lowerfrequency tremor secondary to agerelated changes in the mechanical properties of limbs and muscle. Direct questioning or examination of first degree relatives increases the yield of family history to as high as 96%. Twin studies suggest both hereditary and environmental factors are important in disease expression. Motor control studies show evidence of abnormal production of ballistic movements in a pattern that suggests abnormalities in cerebellar timing. The two most com monly used pharmacological treatments are adrenergic blockers and primidone. Common side effects of these betablocker drugs include bradycardia, fatigue, nausea, diarrhea, rash, impo tence, and depression. Betablockers are contraindicated in patients with congestive heart failure, asthma, thirddegree atrioventricular block, and diabetes. Because of the risk of acute side effects such as vertigo, nausea, and unsteadiness, primidone is usually started at a dose of 25 mg at bedtime and then titrated as tolerated to its effective dose range of 50 to 350 mg daily. Propranolol and primidone combination therapy may be more effective than either agent alone. Botulinum toxin injection in the wrist flexors in patients with prominent hand tremor and into cervical muscles in patients with head tremor may markedly, albeit transiently, reduce the amplitude of the tremor. Adverse effects are relatively rare and may include intracranial hematoma, postoperative seizures, dysarthria, paresthesia, dysequilibrium, headaches, dyspraxia, and wordfinding difficulty. Problems with the stim ulator itself are relatively uncommon but include lead fracture or migration and failure of the impulse generator. About onethird have a positive family history of writing tremor, and a similar number give a history of improvement after ethanol ingestion. Primary writing tremor is not usually associated with the phenomenon of overflow, typically seen in dystonia, and electrophysiological studies suggest it is distinct from both conditions. Accelerometry suggests that the primary writing tremor reflects the normal rhythmic move ment of writing, but the amplitude of the movements is enhanced. The tremor may respond to adrenergic blockade or primidone or anticholinergic medications, but botulinum toxin injections provide the most benefit. Patients may not be aware of the tremor but complain of unsteadiness and vibration or discom fort in the legs that are relieved by leaning against a stationary object, by walking, or by sitting down. Leaning on the arms may precipitate a similar frequency tremor in the arms, and a tremor of the closed jaw has also been reported. A recent study has suggested that coherent highfrequency tremor in the legs may be a normal response to perceived unsteadiness when stand ing still, and that orthostatic tremor may be an exaggeration of this response. Neuropathic Tremor Tremors associated with neuropathy are usually postural and kinetic tremors with a frequency between 3 and 6 cycles per second. The diagnosis is made when a typical tremor affects a person with neuropathy in the absence of other tre morgenic neurological disorders. The pathophysiology of neu ropathic tremor is believed to be disordered feedback control related to abnormal peripheral sensory input. Some patients develop tremor after a peripheral injury, sometimes associated with abnormal posture as well as reflex sympathetic dystrophy or complex regional pain syndrome. Pharmacological treatment is usually disappointing, but some patients respond to betablockers or clonazepam. Cerebellar Tremor the tremor typically associated with cerebellar disease is a slow tremor that is absent during rest but appears and progres sively increases in amplitude with movement, particularly with fine adjustments required for a precise movement. Sitting or standing unsupported may induce a tremor of the trunk and head (titubation). A variant of cerebellar outflow tremor is known as Holmes tremor, or rubral tremor. Rubral tremor results from acquired structural lesions in the ipsilateral cerebellar dentate nucleus and superior cerebellar peduncle. Pharmacological treatment of Holmes tremor is difficult, although some patients respond to levodopa. Hereditary Geniospasm (Chin Tremor) Hereditary geniospasm is characterized by involuntary vertical movement of the tip of the chin with quivering and mouth movements. The disorder is genetically hetero geneous, with linkage to chromosome 9q1321 in some but not all families. Fragile X Premutation Male carriers of the fragile X premutation have been found to have a neurodegenerative syndrome characterized by the onset after age 50 years of kinetic tremor, gait ataxia, executive cognitive dysfunction, parkinsonism, dysautonomia, erectile dysfunction, and peripheral neuropathy (Hagerman and Hager man, 2013). He accurately reported the salient clini cal features of the disease, its pattern of transmission from parent to child, and its dismal prognosis. The disorder is reported in all races, although it is much more common in Scotland and Venezuela and less common in Finland, China, Japan, and black South Africans. Approximately 5% of cases begin in patients younger than 21 years; the juvenile phenotype differs from the adult phenotype, and patients are often misdiagnosed. Clinical Features When clinical illness begins, it does so gradually, and it is best to define a "zone" rather than a time of onset. Patients themselves may be unaware or unconcerned about early cognitive and motor changes. A change in the ability to generate saccadic eye movements and their speed is often the earliest sign. Eventually, a blink or head thrust may be required to initiate saccadic eye move ments. The motor disorder usually begins with clumsiness and fidgetiness that evolves into chorea. The presence and severity of chorea vary markedly from person to person and over time. Some patients, particularly in early stages of the disease, are able to camouflage their chorea by incorporating the involun tary movements into seemingly volitional gestures such as touching their face or adjusting glasses (parakinesia). They may be unable to maintain forced eye closure, hold the mouth open, or protrude the tongue for long periods. With advancing disease, there is progression of bradykinesia, and dystonic movements appear. Ultimately, progressive bradykinesia and intractable falls lead to the wheelchair or bedbound state. Most patients spend the last several years of their lives in nursing home settings and die of complications such as pneu monia and head injury. Mean survival is 17 years, but the natural history varies and is influenced by genetic and envi ronmental factors. The juvenile phenotype differs from the adult phenotype, with prominent parkinsonism and dystonia, even early in the course, and with myoclonus and seizures.
Buy donepezil cheap online
Positron emission tomography studies have shown both postsynaptic and presynaptic dopaminergic changes medications 4 times a day order 5mg donepezil. Some brains of patients of patients with this dystoniaparkinsonism syndrome have been found to have a mosaic pattern of striatal gliosis. In some patients, parkinsonian symptoms are Pathogenesis Many studies have suggested that focal and segmental dysto nia might have a genetic basis (Albanese et al. Approximately 25% of adultonset focal or segmental dystonia patients have a positive family history of dystonia, which would be consistent with an autosomal dominant con dition with low penetrance (Table 96. The pathogenesis of adultonset primary focal or segmental dystonia is unclear, but similar mechanisms to childhood onset primary generalized dystonia are proposed. Several lines of evidence suggest that abnormal central somatosensory processing may lead to insufficient sensorimo tor integration in dystonia. The disorder begins in the first decade of life, usually with an action leg dystonia. The condition then progresses to the fully formed illness that ranges in severity from mild focal to disabling generalized dystonia associated with marked gait difficulty and postural instability with a posi tive pull test. Affected patients may be almost normal in the morning, becoming progressively more disabled over the course of the day, with peak disability late in the evening. Guanosine triphosphate cyclohydrolase 1 is an enzyme involved in the synthesis of tetrahydrobiopterin, a cofactor for tyrosine hydroxylase, the ratelimiting enzyme in the synthesis of levodopa. Another childhoodonset dystonia related to deficient dopaminergic neurotransmission is aromatic acid decarboxy lase deficiency. Dystonia, parkinsonism, oculogyric crises, autonomic symptoms, and progressive neurological impairment begin in childhood. There are deficiencies in central biogenic amines including dopamine, norepinephrine, epinephrine, and serotonin. Dystonia preferentially affects bulbar muscles and progresses over a period of days to weeks but then remains stable. Although sporadic cases have been reported, most cases belong to a small number of families showing dominant inheritance with incomplete penetrance. Over 100 mutations have been already identified and most patients carry at least two muta tions. Many patients present in childhood with symptoms and signs of liver disease ranging from cirrhosis to fulminant liver failure associated with progressive accumulation of copper. Once cirrhosis has developed, extrahepatic copper deposits begin to form, especially in the brain, eyes, and kidneys. Neurological signs usually present during adolescence or early adulthood, but presentations up to age 51 have been reported. Neurological presentations include parkinsonism, postural and kinetic tremor, ataxia, titubation, chorea, seizures, dysarthria, and dys tonia (see Video 96. A fixed stare with a smiling expression and drooling are classic features of the illness but are not seen in all cases. Laboratory studies often show abnormalities in hepatic enzymes, aminoaciduria, low uric acid, and demineralization of bone. Symptoms usually begin before the teenage years and predominantly affect the head, arms, and upper body. Psychiatric features including affective disorder, obsessivecompulsive disorder, substance abuse, anxiety, phobic or panic disorders, and psychosis have been described. Although the neurological disorder clearly relates to harmful effects of intra cellular copper, the precise mechanisms of cell dysfunction and death are not well understood. Traditionally, acute chelation began with dpenicillamine, but more recent treat ment strategies stress somewhat less toxic therapies such as trientine and zinc or ammonium tetrathiomolybdate. The effectiveness of initial decoppering is monitored by serially measuring urine copper excretion and plasma copper levels. Although there may be an acute deterioration associated with the mobilization of copper stores, most patients improve over time. Asymptomatic siblings should be tested for the disease, because timely treatment prevents the illness. Orthotopic liver transplantation is curative but has been used largely in patients with fulminant hepatic failure who have not yet developed significant neurological signs. The response of neu rological symptoms to liver transplantation is not completely understood. Aceruloplasminemia, characterized by anemia, iron overload, diabetes, and neurodegeneration caused by homozygous mutation of the ceruloplasmin gene, may be associated with dystonia and akineticrigid syndrome. Iron chelation with deferiprone has been reported to result in regression of symptoms after 6 months. Most reported posttraumatic dystonias occur in men, reflecting a male preponderance among patients with head injury. Most cases have occurred in children or adolescents who have survived severe head injury. There may be a latent period between the trauma and development of the dystonia from 1 day to 6 years, followed by slow progression of dystonic symptoms. Younger patients tend to have longer latencies than those who are older at the time of the head injury. Focal lesions in the caudate, putamen, or thalamus contralateral to the affected side are usually found on neuroimaging studies. The prognosis of this form of posttraumatic dystonia is poor, with a low rate of spontaneous improve ment. Most cases are refractory to medical therapy, although some may respond to anticholinergic drugs. Botulinum toxin injections may be helpful in the treatment of focal or segmen tal dystonia. Although still somewhat controversial, there is growing evidence that dystonia may also occur after peripheral injury (Jankovic, 2009b). For example, oromandibular dystonia may follow dental surgery or facial and jaw trauma. Limb dystonia has also been reported to occur after peripheral trauma, often in the context of a work or sportsrelated injury. Peripherally induced dystonia tends to manifest with a fixed rather than mobile dystonia and may be associated with complex regional pain syndrome, also known as causalgia or reflex sympathetic dystrophy (Jankovic, 2009b; van Rooijen et al. There are no accepted clinical criteria for this diagnosis, and differ entiation from psychogenic disease may be difficult. It is an autosomal recessive neurode generative disorder presenting in childhood with the insidious onset of dystonia and gait disorder. Rigidity, dysarthria, spas ticity, dementia, retinitis pigmentosa, and optic atrophy develop and progress relentlessly until death in early child hood. Microscopic changes include neuronal loss, gliosis, loss of myelinated fibers, and axonal swellings (spheroids). Because the attacks are often not witnessed and therefore appropriate phenomeno logical categorization is not possible, the less specific term, paroxysmal dyskinesia, is preferred to the alternative term, paroxysmal kinesigenic epilepsy. Patients typically recount that epi sodes are triggered by rapid movement, often in response to an unexpected stimulus such as the telephone ringing. There may be a premonitory sensation in an affected limb, such as limb paresthesia before the onset of the abnormal involuntary movement. Diagnosis depends on careful history taking; because the examination usually shows no abnormalities, typical spells may not be elicited in the examination setting, and neuroimaging and electrophysiological studies are usually normal. Their frequency ranges from several episodes a month to several episodes a day, and their duration is generally between 10 minutes and several hours. They are not precipitated by action but may be triggered by ethanol, caffeine, fatigue, or stress. Secondary paroxysmal dyskinesia has been thought to be rare (Waln and Jankovic, 2015). However, in one series, 26% of paroxysmal dyskinesia cases occurred in the context of another nervous system disease. Underlying etiologies include cerebrovascular disease, trauma, infection, and metabolic encephalopathy.
