

Omeprazole 10 mg sale
Reactions of the innate and acquired immune system including inflammatory processes gastritis diet ��������� generic omeprazole 10 mg otc, blood coagulation reactions, fibrinolysis, and thrombotic processes are intertwined in vivo via multiple molecular and cellular mechanisms. Protein S has direct effects on cells by activating one or more transmembrane receptor tyrosine kinases. Antithrombin can neutralize all coagulation proteases in reactions that are enhanced by heparin and related glycosaminoglycans (see Chap. Antithrombin is key for anticoagulant therapy based on the heparin-stimulated inhibition of thrombin and factor Xa. First, protein S can bind directly to procoagulant factors Xa and Va and thereby inhibit directly the activity of the prothrombinase complex. Following cleavage at the reactive residue in the reactive center loop by a protease, this extended loop is able to partially or completely insert itself into the five-stranded -sheet, forming a very stable six -stranded -sheet. Synthetic pentasaccharides, such as fondaparinux, which are analogues of the naturally occurring sequence, are often termed to be indirect factor Xa inhibitors and have significant clinical utility. For the second mechanistic effect, namely the approximation effect, unfractionated heparin or low-molecular-weight heparins simultaneously bind to antithrombin and the target protease to promote frequent and geometrically productive encounters between protease and inhibitor, thus increasing the reaction rate. The mature antithrombin polypeptide chain contains 432-aminoacid residues after cleavage of a propeptide from a 464-amino-acid-residue precursor. Reactive center defects carry the largest risk of thrombosis, whereas heparin-binding defects are associated with less risk of venous thrombosis (Chap. No protease has yet been identified as the target of the K3 protease inhibitor domain. Curiously, the protease-like domain of protein Z lacks any protease activity because it has mutations at two of the three active site triad residues. Knocking out the protein Z gene in a mouse does not produce a remarkable phenotype unless protein Z deficiency coexists with factor V Leiden, in which case the mouse exhibits a hypercoagulable, prothrombotic state. No association between Chapter 114: Control of Coagulation Reactions 1961 defects in bleeding or thrombosis has been confirmed for this inhibitor. In purified reaction mixtures, protein C inhibitor also efficiently neutralizes thrombin in the presence of thrombomodulin,359 although no studies show that this is a physiologic reaction or that it is associated with thrombosis. Jesty J, Beltrami E, Willems G: Mathematical analysis of a proteolytic positive-feedback loop: Dependence of lag time and enzyme yields on the initial conditions and kinetic parameters. Beltrami E, Jesty J: Mathematical analysis of activation thresholds in enzyme-catalyzed positive feedbacks: Application to the feedbacks of blood coagulation. Kisiel W: Human plasma protein C: Isolation, characterization, and mechanism of activation by alpha-thrombin. Yin T, Miyata T: Dysfunction of protein C anticoagulant system, main genetic risk factor for venous thromboembolism in northeast Asians. Garcia de Frutos P, Fuentes-Prior P, Hurtado B, Sala N: Molecular basis of protein S deficiency. Kimura R, Honda S, Kawasaki T, et al: Protein S-K196E mutation as a genetic risk factor for deep vein thrombosis in Japanese patients. Delvaeye M, Noris M, de Vriese A, et al: Thrombomodulin mutations in atypical hemolytic-uremic syndrome. Ireland H, Kunz G, Kyriakoulis K, et al: Thrombomodulin gene mutations associated with myocardial infarction. Inhibition of activated protein C anticoagulant function without modulation of reaction with proteinase inhibitors. Oganesyan V, Oganesyan N, Terzyan S, et al: the crystal structure of the endothelial protein C receptor and a bound phospholipid. Danese S, Vetrano S, Zhang L, et al: the protein C pathway in tissue inflammation and injury: Pathogenic role and therapeutic implications. Qu D, Wang Y, Song Y, et al: the Ser219->Gly dimorphism of the endothelial protein C receptor contributes to the higher soluble protein levels observed in individuals with the A3 haplotype. Hayashi T, Nakamura H, Okada A, et al: Organization and chromosomal localization of the human endothelial protein C receptor gene. Steinhoff M, Buddenkotte J, Shpacovitch V, et al: Proteinase-activated receptors: Transducers of proteinase-mediated signaling in inflammation and immune response. Zivelin A, Mor-Cohen R, Kovalsky V, et al: Prothrombin 20210G>A is an ancestral prothrombotic mutation that occurred in whites approximately 24,000 years ago. Rodeghiero F, Tosetto A: Activated protein C resistance and factor V Leiden mutation are independent risk factors for venous thromboembolism. Saller F, Kaabache T, Aiach M, et al: the protein S thrombin-sensitive region modulates phospholipid binding and the gamma-carboxyglutamic acid-rich (Gla) domain conformation in a non-specific manner. Riewald M, Ruf W: Protease-activated receptor-1 signaling by activated protein C in cytokine perturbed endothelial cells is distinct from thrombin signaling. Guo H, Singh I, Wang Y, et al: Neuroprotective activities of activated protein C mutant with reduced anticoagulant activity. Wang Y, Thiyagarajan M, Chow N, et al: Differential neuroprotection and risk for bleeding from activated protein C with varying degrees of anticoagulant activity. Guo H, Liu D, Gelbard H, et al: Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Liu D, Cheng T, Guo H, et al: Tissue plasminogen activator neurovascular toxicity is controlled by activated protein C. Wang Y, Zhang Z, Chow N, et al: An activated protein C analog with reduced anticoagulant activity extends the therapeutic window of tissue plasminogen activator for ischemic stroke in rodents. Wang Y, Zhao Z, Chow N, et al: Activated protein C analog promotes neurogenesis and improves neurological outcome after focal ischemic stroke in mice via protease activated receptor 1. Wang Y, Zhao Z, Chow N, et al: Activated protein C analog protects from ischemic stroke and extends the therapeutic window of tissue-type plasminogen activator in aged female mice and hypertensive rats. Structural requirements for thrombin receptor activation reveal mechanism of proteolytic unmasking of agonist function. Xu J, Ji Y, Zhang X, et al: Endogenous activated protein C signaling is critical to protection of mice from lipopolysaccharide induced septic shock. Espana F, Vicente V, Tabernero D, et al: Determination of plasma protein C inhibitor and of two activated protein C-inhibitor complexes in normals and in patients with intravascular coagulation and thrombotic disease. Espana F, Gilabert J, Aznar J, et al: Complexes of activated protein C with alpha 1-antitrypsin in normal pregnancy and in severe preeclampsia. Dahlback B: Purification of human C4b-binding protein and formation of its complex with vitamin K-dependent protein S. Hillarp A, Hessing M, Dahlback B: Protein S binding in relation to the subunit composition of human C4b-binding protein. Uehara H, Shacter E: Auto-oxidation and oligomerization of protein S on the apoptotic cell surface is required for Mer tyrosine kinase-mediated phagocytosis of apoptotic cells. McColl A, Bournazos S, Franz S, et al: Glucocorticoids induce protein S-dependent phagocytosis of apoptotic neutrophils by human macrophages. Gray E, Hogwood J, Mulloy B: the anticoagulant and antithrombotic mechanisms of heparin. Purification and properties of a heparin-dependent inhibitor of thrombin in human plasma. Bolkun L, Galar M, Piszcz J, et al: Plasma concentration of protein Z and protein Z-dependent protease inhibitor in patients with haemophilia A.
Buy cheap omeprazole 10 mg on line
Continuing clozapine with granulocyte colonystimulating factor in patients with neutropenia gastritis peanut butter buy discount omeprazole 20 mg on-line. A non-steady state kinetic evaluation of the mechanism of cortisone-induced granulocytosis. Miki K, Miki M, Nakamura Y, et al: Early-phase neutrophilia in cigarette smokeinduced acute eosinophilic pneumonia. Shoenfeld Y, Tal A, Berliner S, Pinkhas J: Leukocytosis in nonhematological malignancies-A possible tumor-associated marker. Herishanu Y, Rogowski O, Polliack A, Marilus R: Leukocytosis in obese individuals: Possible link in patients with unexplained persistent neutrophilia. Loimaala A, Rontu R, Vuori I, et al: Blood leukocyte count is a risk factor for intimamedia thickening and subclinical carotid atherosclerosis in middle-aged men. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Caramazza D, Caracciolo C, Barone R, et al: Correlation between leukocytosis and thrombosis in Philadelphia-negative chronic myeloproliferative neoplasms. Litos M, Sarris I, Bewley S, et al: White blood cell count as a predictor of the severity of sickle cell disease during pregnancy. Its main function as a phagocytic and bactericidal cell is performed outside the circulation in tissues where microbial invasion takes place. Neutrophil function is traditionally viewed as chemotaxis, phagocytosis, and bacterial killing. Clinical disorders of the neutrophil may arise from impairment of these normal functions. The clinical presentation of a patient who has a qualitative neutrophil abnormality may be similar to that of one who has an antibody, complement, or toll-like receptor disorder. In general, evaluation for phagocyte cell disorders should be initiated among those patients who have at least one of the following clinical features: (1) two or more systemic bacterial infections in a relatively short time period; (2) frequent, serious respiratory infections, such as pneumonia or sinusitis, or otitis media, or lymphadenitis; (3) infections present at unusual sites (liver or brain abscess); and (4) infections associated with unusual pathogens. In the front of the cell is a pseudopodium, referred to as the lamellipodium, that advances before the body of the cell containing the nucleus and the cytoplasmic granules. The lamellipodium undulates or "ruffles" as the neutrophil moves, at a rate of up to 50 m/min. The membrane lipids also flow during locomotion,5 and enhanced cytosolic Ca2+ is observed along the membrane margin. As the cell moves, the cytoplasm behind the lamellipodium streams forward, almost obliterating it. At this point, some granules appear to contact the cell periphery and release granule contents in response to chemotactic agents. A flow of cortical materials, composed particularly of actin filaments, has been proposed to account for chemotaxis as well as other cellular movements. Polarity and movement is orchestrated by the cytoskeleton through signals generated from receptor associated G-proteins in an intricate network that regulates both direction and intensity of movement. In postcapillary venules or in pulmonary capillaries the slow rate of blood flow, further reduced by vessel dilatation at sites of inflammation, permits a loose and somewhat transient adhesion referred to as "tethering," and results in the rolling of the neutrophil along the endothelium. Cinemicrophotographic observation of granule lysis of a chicken neutrophil following phagocytosis of zymosan particles. Note the lysis of the cytoplasmic granule (G) against one of two ingested zymosan particles (Z). As with locomotion, phagocytosis results in Ca2+ being released in the vicinity of the active membranes. When dissolution of the lamellipodium occurs, the interior contents of the cell are allowed to contact the cell membrane. Fusion of membranes is a common feature of (1) ingestion, where pseudopodia fuse; (2) degranulation, where granules fuse with the phagosome; and possibly (3) locomotion, where some granules may fuse with the plasma membrane. Pseudopodia form whether neutrophils are suspended in liquid medium or are attached to a surface, but the cell can only move translationally when fixed to a surface; thus it crawls but does not swim. Thus, the formation of pseudopodia, membrane fusion, and membrane adhesiveness are all characteristics associated with the functional responses of neutrophils. L-selectin is constitutively present on neutrophils and its binding capacity is rapid and transiently increased after neutrophil activation, possibly via receptor oligomerization. Initially neutrophils appear at sites on the endothelium adjacent to the site of inflammation. E- and P-selectin serve as counterreceptors for the neutrophil P-selectin glycoprotein ligand-1. The 2 integrins mediate tight adhesion and arrest of the leukocytes in cooperation with the selectins. The extracellular domains of unactivated integrins are in a bent position and not able to bind ligands. This changes the conformation of the extracellular domains from the bent to the open state, thereby permitting binding to ligands, and in so doing transmit signals from outside to inside. A minority of neutrophils exit by a transcellular route through so-called endothelial cups. The surface of neutrophils is highly dynamic as a result of the incorporation of membrane from intracellular vesicles and granules, a process that is known to add significantly to the total cell surface measured by an increase in electric capacitance. This enhances the ability of neutrophils to respond to the signals presented by endothelial cells or present in the extravascular tissue. Both growth factor receptors are important for myeloid development, and play an important role in enhancing neutrophil function and gene transcription in mature neutrophils. Lipid rafts are important, but elusive structures that facilitate signal transduction leading to phagocytosis by promoting several membrane protein interactions. Initially the rafts were conceptionally associated with caveolae, which are structures identified on endothelial cells and thought to be important for transendothelial cell traffic. The caveolae were identified by their high content of cholesterol lipids and the presence of the structural protein, caveolin. Rafts were subsequently identified on neutrophils, but these cells are devoid of caveolin. Other membrane protein receptors that are not normally associated with rafts may change their conformation and subsequently associate with rafts upon binding their ligands. When Paul Ehrlich introduced aniline dyes in histochemistry and discovered the different subsets of leukocytes, the neutrophil granules were divided into those that took up the azure dye, the azurophilic granules, and the others, the specific granules. Yet nature has provided a beautiful setting that allows the neutrophil to fine tune its response to a specific task. A priori, there would be two reasons for having different subsets of granules: One would be to ensure that proteins, which cannot coexist, are segregated; that is, protease-sensitive proteins are separated from proteases. The other reason would be to have proteins whose service is needed at one time separated from proteins whose service is needed at a different time. Secretory vesicles of neutrophils are specialized endocytosis vesicles that are formed in the final stages of neutrophil maturation in the marrow. Albumin thus serves as a marker for secretory vesicles and has allowed the identification of these as small intracellular vesicles that are scattered throughout the cytoplasm of neutrophils as is true for neutrophil granules. The plasma proteins inside secretory vesicles show no sign of degradation, thus no fusion takes place with lysosomal structures. Among the peroxidase-positive granules, subsets can be identified that are rich in defensins as well as some that are not. Peroxidase-negative granules can be divided into three subsets based on the distribution of the two marker proteins lactoferrin and gelatinase: granules that contain lactoferrin, but no gelatinase (15 percent of peroxidase-negative granules), granules that contain both proteins (60 percent), and granules that are rich in gelatinase, but low (or absent) in lactoferrin (25 percent). Following neutrophil stimulation, gelatinase granules are exocytosed to a larger extent than granules containing both lactoferrin and gelatinase, and these are more readily mobilized than granules containing lactoferrin but lacking gelatinase. Granule proteins are synthesized during myelopoiesis from myeloblasts to band cells and segmented neutrophils in the marrow.
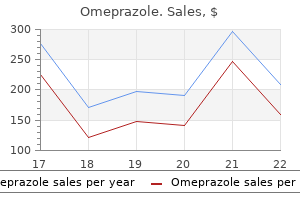
Order omeprazole
Hypogammaglobulinemia of variable degree can be observed gastritis and stress order omeprazole 20mg, and immunizations result in short-lived antibody responses and impaired class switch. Immunoglobulin replacement therapy and antibiotic prophylaxis may reduce the incidence of infections. Warts are resistant to local therapy and need to be monitored for neoplastic transformation. Disease-specific abnormalities involving growth and development, the central nervous system, and the skin provide useful diagnostic clues. The genes responsible for these syndromes protect human genome integrity by contributing to the complex task of double-strand break repair. Together with the proteins associated with Fanconi anemia, the gene products of the chromosomal instability syndromes form or regulate a large protein complex that is active in the surveillance and maintenance of genomic integrity. The number of circulating lymphocytes is often reduced, and proliferation in response to mitogens is variably depressed. Spontaneous cytogenetic abnormalities include chromosomal breaks, translocations, rearrangements, and inversions; these defects increase following in vitro exposure to radiation. The thymus is often small, showing marked paucity of thymocytes and absence of Hassall corpuscles. The most consistent laboratory abnormality, an elevation of serum -fetoprotein, is diagnostic in adults and children older than age 8 months as it is not observed in the other chromosomal instability syndromes. Eventually, involuntary movements become a major handicap and the child may require a wheelchair by the end of the first decade of life. Cortical cerebellar degeneration involves primarily Purkinje and granular cells; progressive changes to the central nervous system also occur. Because of hypersensitivity to radiation and radiomimetic/alkylating agents, tumor therapy is limited. Leukemia and non-Hodgkin lymphoma predominate during the first two decades; later, carcinoma affecting the colon, skin, and breast are common. Because of increased radiation sensitivity, exposure to any form of irradiation should be restricted. Overall, these disorders are characterized by increased susceptibility to severe viral infections that in some cases is associated with defects of hair and skin pigmentation, and neurologic problems. This results in life-threatening manifestations, characterized by fever, hepatosplenomegaly, marrow infiltration and pancytopenia, and severe neurologic manifestations (Chap. Treatment of active disease should focus on controlling or eliminating possible triggers (infections in particular), blocking T-cell activation, and stopping the hyperinflammatory cytokine response. To this purpose, antimicrobials, etoposide, immune suppression (antithymocyte globulin), cyclosporine, and dexamethasone are commonly used. Patients should be monitored carefully for reactivation of the disease, especially in the central nervous system. Use of myeloablative conditioning regimen is associated with high transplantation-related mortality. Considering that partial chimerism is enough to achieve disease control,244 reduced intensity conditioning is being increasingly used, with promising results. Neurologic symptoms, including seizures and decreased level of consciousness, may lead to long-term disability. Hemophagocytosis can be observed in the marrow, lymph nodes, and cerebrospinal fluid, which often shows abundant mononuclear cells and increased proteins, even in the absence of overt neurologic symptoms. Most lymphomas are of B-cell origin, and approximately half are of the Burkitt type. Persistent dysgammaglobulinemia, with low IgG and low to increased levels of IgM, is common among survivors. Bruises are common and reflect deficiency of the platelet specific granules responsible for secondary aggregation (Chap. Both bacterial and viral infections may trigger the life-threatening "accelerated phase" of the disease, characterized by high fever, hepatosplenomegaly, coagulation abnormalities, increase of liver enzymes and bilirubin (with possible jaundice), edema, and neurologic symptoms, with seizures, ataxia, cranial nerve palsies, and peripheral neuropathy. Light microscopy examination of hair reveals large and evenly distributed granules of melanin. Treatment requires control of infections, and immunosuppressive intervention during the accelerated phase. The mortality rate is particularly high (96 percent) in patients who present with fulminant infectious mononucleosis. Use of myeloablative conditioning is associated with high mortality and toxicity rate, but good results have been reported with reduced intensity conditioning. Prominent hypopigmentation is a result of large clumps of melanin in the hair shafts. Reduced platelet-dense granules and impaired platelet degranulation are responsible for increased susceptibility to bleeding. Susceptibility to recurrent meningitis caused by Neisseria meningitidis is discussed in "Genetically Determined Deficiencies of the Complement System" below. Use of antimicrobic prophylaxis is important to prevent invasive pyogenic infections, especially in childhood. Substitution therapy with immunoglobulins may be beneficial in patients with impaired antibody responses. Because of its progression to myelodysplastic syndrome or acute myeloid leukemia, mortality can be as high as 28 percent. Affected individuals have either a mild clinical course, characterized by selective susceptibility to mycobacterial infections, or are asymptomatic. Mutations in the classical pathway (C1q, C1r/C1s, C4, C2, and C3) result in pyogenic infections and autoimmune diseases. Mutations affecting the alternative pathway (factors B, D, properidin) result in meningococcal and pneumococcal sepsis. All other complement-component deficiencies have an autosomal recessive mode of inheritance with the exception of properidin deficiency, which is X-linked. To pinpoint the specific complement component deficiency, immunochemical tests using component specific antibodies or functional assays using in vitro reconstitution of the hemolytic function are recommended. Autoimmune disorders are treated symptomatically, using the same immunosuppressive agents and antiinflammatory medications as those used in the general population. Management of angioedema has been revolutionized by C1 esterase inhibitor concentrate (Cinryze, Berinert), which is most effective for the treatment of acute attacks and by the kallikrein inhibitor Kalbitor (Dyax) and the bradykinin receptor antagonist Firazyr (Shire). Plebani A, Soresina A, Rondelli R, et al: Clinical, immunological, and molecular analysis in a large cohort of patients with X-linked agammaglobulinemia: An Italian multicenter study. Etzioni A, Ben-Barak A, Peron S, et al: Ataxia-telangiectasia in twins presenting as autosomal recessive hyper-immunoglobulin M syndrome. Seyama K, Kobayashi R, Hasle H, et al: Parvovirus B19-induced anemia as the presenting manifestation of X-linked hyper-IgM syndrome. Levy J, Espanol-Boren T, Thomas C, et al: Clinical spectrum of X-linked hyper-IgM syndrome. Angulo I, Vadas O, Garcon F, et al: Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Salzer E, Santos-Valente E, Klaver S, et al: B-cell deficiency and severe autoimmunity caused by deficiency of protein kinase C delta. Kanoh T, Mizumoto T, Yasuda N, et al: Selective IgA deficiency in Japanese blood donors: Frequency and statistical analysis. Fischer A, Le Deist F, Hacein-Bey-Abina S, et al: Severe combined immunodeficiency. Pannicke U, Honig M, Hess I, et al: Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Neven B, Leroy S, Decaluwe H, et al: Long-term outcome after hematopoietic stem cell transplantation of a single-center cohort of 90 patients with severe combined immunodeficiency. Yeganeh M, Heidarzade M, Pourpak Z, et al: Severe combined immunodeficiency: A cohort of 40 patients. Dror Y, Grunebaum E, Hitzler J, et al: Purine nucleoside phosphorylase deficiency associated with a dysplastic marrow morphology. Buck D, Malivert L, de Chasseval R, et al: Cernunnos, a novel nonhomologous endjoining factor, is mutated in human immunodeficiency with microcephaly. Titman P, Pink E, Skucek E, et al: Cognitive and behavioral abnormalities in children after hematopoietic stem cell transplantation for severe congenital immunodeficiencies.
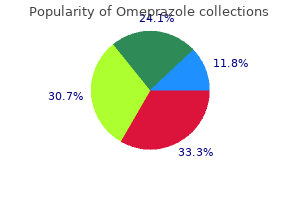
Order 10mg omeprazole
Mononeuritis multiplex is a particularly characteristic feature that sometimes leads to rapid onset of a peripheral neuropathy chronic gastritis years 20mg omeprazole free shipping, which can be permanent, causing considerable morbidity. Cardiac disease with valvular damage or endomyocarditis is one of the most serious complications. There are case reports of family clusters, but in most cases there is no evidence of an inherited cause. In most cases, patients go into remission with highdose oral glucocorticoids, although guidelines recommend the use of other immunosuppressants, such as cyclophosphamide or azathioprine, to induce remission where there is evidence of life-threatening organ involvement. Relapse is not uncommon and this, together with the adverse effects of medication, result in significant morbidity. The widely used American College of Rheumatology criteria were for classification purposes rather than to aid diagnosis. Because there was no comparison with asthma or other eosinophilic disorders, the criteria lack specificity. The diagnostic hallmark is evidence of an eosinophilic vasculitis with granuloma on biopsy, but in many cases it is not possible to obtain tissue and the diagnosis is based on clinical assessment. To identify novel biomarkers, a retrospective audit of patients who had been investigated at a tertiary center for an unexplained eosinophilia were studied. Eosinophiluria and Eosinophilorrachia the urinary excretion of eosinophils is seen in several inflammatory disorders of the kidney but most often in urinary tract infection or acute interstitial nephritis. Cerebrospinal fluid eosinophilia may occur with infection, shunts, and allergic reactions involving the meninges. Egesten A, Calafat J, Janssen H, et al: Granules of human eosinophilic leukocytes and their mobilization. Persson T, Calafat J, Janssen H, et al: Specific granules of human eosinophils have lysosomal characteristics: Presence of lysosome-associated membrane proteins and acidification upon cellular activation. Piecemeal degranulation of specific granules and distribution of Charcot-Leyden crystal protein. Clark K, Simson L, Newcombe N, et al: Eosinophil degranulation in the allergic lung of mice primarily occurs in the airway lumen. Munday J, Kerr S, Ni J, et al: Identification, characterization and leucocyte expression of Siglec-10, a novel human sialic acid-binding receptor. Eosinophilic Fasciitis this rare syndrome may occur at any age in either sex and is characterized by stiffness, pain, and swelling of the arms, forearms, thighs, legs, hands, and feet in descending order of frequency. A biopsy, usually required for the diagnosis, shows inflammation, edema, thickening, and fibrosis of the involved fascia. Aplastic anemia, isolated cytopenias, pernicious anemia, and acute myelogenous leukemia have been associated with eosinophilic fasciitis and in the late 1980s a series of cases was described in association with ingestion of a particular batch of l-tryptophan. Takatsu K, Kouro T, Nagai Y: Interleukin 5 in the link between the innate and acquired immune response. Mjosberg J, Eidsmo L: Update on innate lymphoid cells in atopic and non-atopic inflammation in the airways and skin. Letuve S, Druilhe A, Grandsaigne M, et al: Involvement of caspases and of mitochondria in Fas ligation-induced eosinophil apoptosis: Modulation by interleukin-5 and interferon-gamma. Letuve S, Druilhe A, Grandsaigne M, et al: Critical role of mitochondria, but not caspases, during glucocorticosteroid-induced human eosinophil apoptosis. Phipps S, Ying S, Wangoo A, et al: the relationship between allergen-induced tissue eosinophilia and markers of repair and remodeling in human atopic skin. Persson C, Uller L: Theirs but to die and do: Primary lysis of eosinophils and free eosinophil granules in asthma. Kaneko M, Horie S, Kato M, et al: A crucial role for beta 2 integrin in the activation of eosinophils stimulated by IgG. Kovacs I, Horvath M, Kovacs T, et al: Comparison of proton channel, phagocyte oxidase, and respiratory burst levels between human eosinophil and neutrophil granulocytes. Kimura I, Moritani Y, Tanizaki Y: Basophils in bronchial asthma with reference to reagin-type allergy. Juhlin L, Michaelsson G: A new syndrome characterised by absence of eosinophils and basophils. Cogan E, Schandene L, Crusiaux A, et al: Brief report: Clonal proliferation of type 2 helper T cells in a man with the hypereosinophilic syndrome. Roufosse F, Schandene L, Sibille C, et al: Clonal Th2 lymphocytes in patients with the idiopathic hypereosinophilic syndrome. Roufosse F, Cogan E, Goldman M: Recent advances in pathogenesis and management of hypereosinophilic syndromes. Helbig G, Soja A, Bartkowska-Chrobok A, et al: Chronic eosinophilic leukemia-not otherwise specified has a poor prognosis with unresponsiveness to conventional treatment and high risk of acute transformation. Pitini V, Arrigo C, Azzarello D, et al: Serum concentration of cardiac Troponin T in patients with hypereosinophilic syndrome treated with imatinib is predictive of adverse outcomes. Sable-Fourtassou R, Cohen P, Mahr A, et al: Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Comarmond C, Pagnoux C, Khellaf M, et al: Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): Clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Mouthon L, Dunogue B, Guillevin L: Diagnosis and classification of eosinophilic granulomatosis with polyangiitis (formerly named Churg-Strauss syndrome). Khoury P, Zagallo P, Talar-Williams C, et al: Serum biomarkers are similar in ChurgStrauss syndrome and hypereosinophilic syndrome. In humans, basophils are the least frequent of the three granulocytes, typically accounting for less than 0. Basophils circulate as mature cells and can be recruited into tissues, particularly at sites of immunologic or inflammatory responses, but they ordinarily do not reside in tissues. By contrast, mast cells typically are derived from blood precursors that lack many of the characteristic features of the mature cells and complete their maturation in the tissues. Mast cells are particularly abundant near blood vessels and nerves and in connective tissues beneath surfaces that are exposed to the external environment, such as the skin, gastrointestinal and urogenital tracts, and respiratory system. Tissue mast cell numbers can increase at sites of parasite infection or in association with certain chronic allergic diseases or other forms of pathology, by recruitment and local maturation of blood precursors and by proliferation of resident mast cells. Accordingly, mast cells and basophils have long been regarded as important effector cells in asthma, hay fever, and other allergic disorders. However, several lines of evidence indicate mast cells and basophils also contribute to protective host responses associated with IgE production, especially those directed against parasites. In mice, mast cells can enhance innate and acquired (IgE-dependent) defense against animal venoms and also can contribute to host defense in innate immune responses to certain bacterial infections. Mast cells and basophils also may express positive and negative immunoregulatory functions through cytokine production and other mechanisms. Although a variety of systemic disorders are associated with changes in the numbers of blood basophils and many pathologic processes can be associated with changes in the numbers of tissue mast cells, patients with primary deficiencies in basophils appear to be exceedingly rare (if they exist at all). Increased numbers of basophils may be present in association with myeloproliferative neoplasms and several forms of myeloid leukemia. Increased numbers of basophils, sometimes to levels of 20 to 90 percent of blood leukocytes, occur in virtually all patients with chronic myelogenous leukemia. The basophils associated with both chronic myelogenous and acute myeloid leukemias are themselves part of the neoplastic clone. The management of patients with "basophilic leukemia" can be complicated by shock as a result of massive release of histamine and other mediators in association with acute cytolysis. Disorders of mast cell hyperplasia/neoplasia include solitary mastocytomas, the pathogenesis of which is uncertain, the spectrum of disorders encompassed in the term mastocytosis, in which significantly increased numbers of mast cells occur in the skin and/or other organs, and mast cell leukemia. The most common form of mastocytosis, indolent systemic mastocytosis, typically presents with urticaria pigmentosa involving the skin, although other organs may be involved. Patients with indolent systemic mastocytosis have the best prognosis and can expect a normal life span. Patients with aggressive systemic mastocytosis have a guarded prognosis because of complications arising from rapid increases in tissue mast cell numbers. Patients with mast cell leukemia, who often present with large numbers of immature mast cells in the blood at the time of diagnosis, have a fulminant and rapidly fatal course. Much evidence indicates that basophils share a common precursor with other granulocytes and monocytes. At least some mast cells can proliferate in the tissues during a variety of inflammatory or reparative processes. At least four mechanisms may account for phenotypic variation in mast cell populations: (1) factors promoting branching within the mast cell lineage; (2) factors influencing differentiation and maturation (within a single pathway or, if they occur, within multiple pathways); (3) factors modulating mast cell function; and (4) factors influencing local concentrations of exogenous substances not derived from mast cells but taken up and stored in mast cell granules. Of these four mechanisms, experimental evidence has been obtained for all but the first.
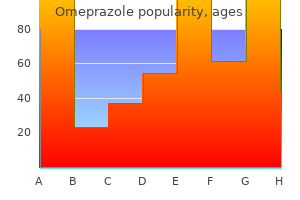
Diseases
- Cryophobia
- Thompson Baraitser syndrome
- Lymphangiomatosis, pulmonary
- Ovarian carcinosarcoma
- Lymphedema, congenital
- Myoclonus progressive epilepsy of Unverricht and Lundborg
- X chromosome, trisomy Xpter Xq13
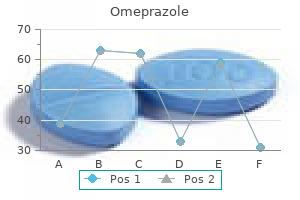
Omeprazole 40 mg mastercard
Hemoglobin or hematocrit levels are followed as safety (not therapeutic) targets gastritis enteritis cheap 20 mg omeprazole visa, to prevent symptomatic anemia. Usually, the hemoglobin should not fall below 10 to 11 g/dL, but the baseline value and the age and clinical condition of the patient are also considered. The therapeutic target is a serum ferritin near 15 ng/mL, which is close to the lower limit of normal and associated with tissue iron depletion but usually not Therapy anemia. Treatment is also guided by plasma (or serum) porphyrin levels, which are more convenient to measure repeatedly than urine porphyrins, and fall more slowly than the serum ferritin. Plasma porphyrins usually decline from initial levels of 10 to 25 mcg/dL during treatment, to below the upper limit of normal (~1 mcg/dL) within weeks after phlebotomies are completed. However, relapses may occur, especially in patients who resume use of alcohol, and are treated by another course of phlebotomies. It is also advisable to follow porphyrin levels and reinstitute phlebotomies promptly if porphyrin levels begin to rise. Liver imaging and a serum -fetoprotein determination should be repeated as screening for hepatocellular carcinoma. However, this treatment is preferred at some centers because it is more convenient and much less expensive. A recent prospective study found that time to biochemical remission with low-dose hydroxychloroquine was comparable to that with phlebotomy. These 4-aminoquinolines are not effective in other porphyrias, and do not mobilize all types of porphyrins from liver and other tissues. It was suggested that mobilization of hepatic iron may be important,302,312,313 but serum ferritin concentrations do not change significantly during treatment. Most likely, these drugs colocalize with excess porphyrins in lysosomes and other intracellular organelles and promote their release by a process that involves transient cell damage. Second, there is some evidence that treatment of hepatitis C may be more effective after iron reduction. Hydroxychloroquine may be an option during treatment with interferon and ribavirin, but initial worsening of liver function tests, even with a low-dose regimen, may cause concern. Studies are needed with newer and more effective agents for hepatitis C as these become available. Erythropoietin administration can correct anemia, mobilize iron, and support phlebotomy in many cases. The level of plasma porphyrins are often especially high in these patients, and should be assessed prior to surgery, because there may be some risk of skin and peritoneal burns from exposure to operating room lights. Retrovirus-mediated gene transfer can correct porphyria in cell lines from patients with this disease, which suggests that gene therapy may be applicable in the future. Schultz J: Ein fall von pemphigus, kompliziert durch lepra visceralis, in Medicine. Cam C, Nigogosyan G: Acquired toxic porphyria cutanea tarda due to hexachlorobenzene. Sassa S, Kappas A: Genetic, metabolic and biochemical aspects of the porphyrias, in Advances in Human Genetics, edited by Harris H, Hirschhorn K, p 121. Podvinec M, Handschin C, Looser R, et al: Identification of the xenosensors regulating human 5-aminolevulinate synthase. Furuyama K, Fujita H, Nagai T, et al: Pyridoxine refractory X-linked sideroblastic anemia caused by a point mutation in the erythroid 5-aminolevulinate synthase gene. Tsukamoto I, Yoshinaga T, Sano S: the role of zinc with special reference to the essential thiol groups in delta-aminolevulinic acid dehydratase of bovine liver. Profound inhibition of delta-aminolevulinic acid dehydratase activity by succinylacetone. Lindblad B, Lindstedt S, Steen G: On the enzymic defects in hereditary tyrosinemia. Chretien S, Dubart A, Beaupain D, et al: Alternative transcription and splicing of the human porphobilinogen deaminase gene result either in tissue-specific or in housekeeping expression. Grandchamp B, De Verneuil H, Beaumont C, et al: Tissue specific expression of porphobilinogen deaminase. Romana M, Dubart A, Beaupain D, et al: Structure of the gene for human uroporphyrinogen decarboxylase. Cacheux V, Martasek P, Fougerousse F, et al: Localization of the human coproporphyrinogen oxidase gene to chromosome band 3q12. Takahashi S, Furuyama K, Kobayashi A, et al: Cloning of a coproporphyrinogen oxidase promoter regulatory element binding protein. Wada O, Sassa S, Takaku F, et al: Different responses of the hepatic and erythropoietic delta-aminolevulinic acid synthetase of mice. Hallai N, Anstey A, Mendelsohn S, et al: Pregnancy in a patient with congenital erythropoietic porphyria. Dupuis-Girod S, Akkari V, Ged C, et al: Successful match-unrelated donor bone marrow transplantation for congenital erythropoietic porphyria (Gunther disease). Nakahashi Y, Fujita H, Taketani S, et al: the molecular defect of ferrochelatase in a patient with erythropoietic protoporphyria. Gouya L, Puy H, Lamoril J, et al: Inheritance in erythropoietic protoporphyria: A common wild-type ferrochelatase allelic variant with low expression accounts for clinical manifestation. Shirota T, Yamamoto H, Hayashi S, et al: Myelodysplastic syndrome terminating in erythropoietic protoporphyria after 15 years of aplastic anemia. Different rates of disappearance of protoporphyrin from the erythrocytes, both in vivo and in vitro. Sandberg S, Brun A, Hovding G, et al: Effect of zinc on protoporphyrin induced photohaemolysis. Bloomer J, Wang Y, Singhal A, et al: Molecular studies of liver disease in erythropoietic protoporphyria. Delaby C, Lyoumi S, Ducamp S, et al: Excessive erythrocyte ppix influences the hematologic status and iron metabolism in patients with dominant erythropoietic protoporphyria. Turnbull A, Baker H, Vernon-Roberts B, et al: Iron metabolism in porphyria cutanea tarda and in erythropoietic protoporphyria. Fontanellas A, Mazurier F, Landry M, et al: Reversion of hepatobiliary alterations by bone marrow transplantation in a murine model of erythropoietic protoporphyria. Hassoun A, Verstraeten L, Mercelis R, et al: Biochemical diagnosis of an hereditary aminolaevulinate dehydratase deficiency in a 63-year-old man. Plewinska M, Thunell S, Holmberg L, et al: Delta-aminolevulinate dehydratase deficient porphyria: Identification of the molecular lesions in a severely affected homozygote. Akagi R, Nishitani C, Harigae H, et al: Molecular analysis of delta-aminolevulinate dehydratase deficiency in a patient with an unusual late-onset porphyria. Akagi R, Yasui Y, Harper P, et al: A novel mutation of delta-aminolaevulinate dehydratase in a healthy child with 12% erythrocyte enzyme activity. Akagi R, Inoue R, Muranaka S, et al: Dual gene defects involving delta-aminolaevulinate dehydratase and coproporphyrinogen oxidase in a porphyria patient. Thunell S, Henrichson A, Floderus Y, et al: Liver transplantation in a boy with acute porphyria due to aminolaevulinate dehydratase deficiency. Shimizu Y, Ida S, Naruto H, et al: Excretion of porphyrins in urine and bile after the administration of delta-aminolevulinic acid. Doss M, von Tiepermann R, Schneider J, et al: New type of hepatic porphyria with porphobilinogen synthase defect and intermittent acute clinical manifestation. Gross U, Sassa S, Jacob K, et al: 5-Aminolevulinic acid dehydratase deficiency porphyria: A twenty-year clinical and biochemical follow-up. Thunell S, Holmberg L, Lundgren J: Aminolaevulinate dehydratase porphyria in infancy. Mercelis R, Hassoun A, Verstraeten L, et al: Porphyric neuropathy and hereditary delta-aminolevulinic acid dehydratase deficiency in an adult. Fujita H, Sato K, Sano S: Increase in the amount of erythrocyte delta-aminolevulinic acid dehydratase in workers with moderate lead exposure. Sassa S, Fujita H, Kappas A: Succinylacetone and delta-aminolevulinic acid dehydratase in hereditary tyrosinemia: Immunochemical study of the enzyme.
Discount omeprazole 20mg fast delivery
In this group gastritis upper gi bleed buy generic omeprazole 10 mg on-line, occasional patients experience increases in monoclonal protein concentration of up to 50 percent of their initial diagnostic value. However, these patients restabilize and do not develop signs of myeloma, macroglobulinemia, amyloidosis, or lymphoma. About half of patients die of an unrelated cause over the 25to 30-year period of observation. The remaining 25 percent of patients develop a plasmacytoma, myeloma, amyloidosis, macroglobulinemia, lymphoma, or chronic lymphocytic leukemia over several decades of observation. The occurrence of a lymphoma or myeloma in the latter group of patients continues to increase slowly without reaching a plateau. Evolution to a progressive clonal B-cell disorder has been observed Chapter 106: Essential Monoclonal Gammopathy 1727 more than 25 years after the diagnosis of monoclonal gammopathy. The actuarial risk of progressing to a clonal B-cell malignancy for all classes of monoclonal protein is approximately 0. Neither the plasma cell gene-expression profile nor the cell population cytogenetic findings are sufficiently specific to predict progression from a stable to an unstable clone based on current studies. In rare patients, the monoclonal protein appears transiently in relation to a disease. One of the most subtle interfaces is between essential monoclonal gammopathy and smoldering myeloma (Chap. In the latter, the marrow plasma cell concentration is between 10 and 20 percent or the monoclonal protein concentration is greater than 3 g/dL or both. Although at this time treatment is not recommended for smoldering myeloma until progression, calls for clinical trials to assess whether early treatment may improve outcome have been made. Therapy may be indicated, however, if the monoclonal protein interferes with the vital function of a normal plasma or tissue constituent, induces kidney disease, or is associated with a disabling neuropathy. A better understanding and an ability to identify the somatic mutations that underlie the evolution of monoclonal gammopathy to a progressive and potentially fatal B-cell neoplasm may permit the application of therapy at an earlier time when curability or sustained remissions would be more frequent. Axelsson U, Bachmann R, Hallen J: Frequency of pathological proteins (M-components) in 6995 sera from an adult population. Iwanaga M, Tagawa M, Tsukasaki K, et al: Prevalence of monoclonal gammopathy of undetermined significance: Study of 52,802 persons in Nagasaki City, Japan. Prevalence of monoclonal gammopathy of undetermined significance in Asia: A viewpoint from Nagasaki atomic bomb survivors. Bizzaro N, Pasini P: Familial occurrence of multiple myeloma and monoclonal gammopathy of undetermined significance in siblings. Pasqualetti P, Collacciani A, Casole R: Risk of monoclonal gammopathy of undetermined significance. Brousseau M, Leleu X, Gerard J, et al: Hyperdiploidy is a common finding in monoclonal gammopathy of undetermined significance and monosomy 13 is restricted to these hyperdiploid patients. Avet-Loiseau H, Li J-Y, Morineau N: Monosomy 13 is associated with the transition of monoclonal gammopathy of undetermined significance to multiple myeloma. Lloveras E, Sole F, Florensa L, et al: Contribution of cytogenetics and in situ hybridization to the study of monoclonal gammopathy of undetermined significance. Zhan F, Hardin J, Kordesmeier B, et al: Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Transplantation of the paraprotein-producing clone from old to young 57B1/KaLwRij mice. George G, Gilburd B, Schoenfeld Y: the emerging concept of pathogenic natural antibodies. Milla F, Oriol A, Aguilar J, et al: Usefulness and reproducibility of cytomorphic evaluations to differentiate myeloma from monoclonal gammopathies of unknown significance. Kinoshita K, Nagai H, Murate T, et al: IgD monoclonal gammopathy of undetermined significance. Jensen K, Jensen B, Olesen H: Three M-components in serum from an apparently healthy person. Jagannath S: Value of serum free light chain testing for the diagnosis and monitoring of monoclonal gammopathies in hematology. Mangiacavalli S, Cocito F, Pochintesta L, et al: Monoclonal gammopathy of undetermined significance: A new proposal of workup. Yujiri T, Nakamura Y, Oota I, et al: Acquired von Willebrand syndrome associated with monoclonal gammopathy of undetermined significance. Wasada T, Egueli Y, Takayama S, et al: Insulin autoimmune syndrome associated with benign monoclonal gammopathy. Disdier P, Swiader L, Aillaud M-F, et al: Ig M monoclonal gammopathy, lymphoid proliferations and lupus anticoagulant. Gavarotti P, Fortina F, Costa D, et al: Benign monoclonal gammopathy presenting with severe renal failure. Maes B, Vanwalleghem J, Kuypers D, et al: IgA antiglomerular basement membrane disease associated with bronchial carcinoma and monoclonal gammopathy. Hashimoto T, Arakawa K, Ohta Y, et al: Acquired Fanconi syndrome with osteomalacia secondary to monoclonal gammopathy of undetermined significance. Paladini I, Pieretti G, Giuntoli M, et al: Crystalline corneal deposits in monoclonal gammopathy: In-vivo confocal microscopy. Ellie E, Vital A, Steck A, et al: Neuropathy associated with "benign" anti-myelinassociated glycoprotein IgM gammopathy: Clinical, immunological, neurophysiological pathological findings and response to treatment in 33 cases. Di Troia A, Carpo M, Meucci N, et al: Clinical features and anti-neural reactivity in neuropathy associated with IgG monoclonal gammopathy of undetermined significance. Nicholas G, Maisonobe T, Le Forestier N, et al: Proposed revised electrophysiological criteria for chronic inflammatory demyelinating polyradiculopathy. Vital A, Nedelec-Ciceri C, Vital C: Presence of crystalline inclusions in the peripheral nerve of a patient with IgA lambda monoclonal gammopathy of undetermined significance. Latov N: Pathogenesis and therapy of neuropathies associated with monoclonal gammopathies. Sghirlanzoni A, Solari A, Ciano C: Chronic inflammatory demyelinating polyradiculopathy: Long-term course and treatment of 60 patients. Burner E, Swahlen A, Cruchaud A: Nonmalignant monoclonal immunoglobulinemia, pernicious anemia, and gastric carcinoma: A model of immunologic dysfunction. Ilfeld D, Barzilay J, Vana D, et al: IgG monoclonal gammopathy in four patients with polymyalgia rheumatica [letter]. Wallach D, Carado Y, Foldes C, Cottennot F: Dermatomyositis and monoclonal gammopathy. Johnsson V, Svendsen B, Vostrup S, et al: Multiple autoimmune manifestations in monoclonal gammopathy of undetermined significance and chronic lymphocytic leukemia. Samochocki Z, Szudzinski A: Gangrenous pyoderma in monoclonal IgA gammopathy and functional disorders of T lymphocytes. Paul C, Fermaud J-P, Flageul B, et al: Hyperkeratotic spicules and monoclonal gammopathy. Rodic P, Pavlovic S, Kostic T et al: Gammopathy and B lymphocyte clonality in patients with Gaucher type I disease. Hamazaaki K, Baba M, Hasegawa H, et al: Chronic hepatitis associated with monoclonal gammopathy of undetermined significance. Dizdar O, Erman M, Cankurtaran M, et al: Lower bone mineral density in geriatric patients with monoclonal gammopathy of undetermined significance. Landgren O, Mailankody S: Update on second primary malignancies in multiple myeloma: A focused review. Shoenfeld Y, Berliner S, Ayalone A, et al: Monoclonal gammopathy in patients with chronic and acute myeloid leukemia. Tosato F, Fossaluzza V, Rossi P, et al: Monoclonal gammopathy of undetermined significance in a case of primary thrombocythemia. Economopoulos T, Economidou J, Papageorgiou E, et al: Monoclonal gammopathy in chronic myeloproliferative disorders.
Discount 40mg omeprazole with visa
The two identical F(ab) pieces contain the entire light chain and the aminoterminal portion of the heavy chain diet gastritis erosif buy 20mg omeprazole otc. The Fc-regions also contain a binding epitope for the neonatal Fc receptor (FcRn), responsible for the extended half-life, placental transport, and bidirectional transport of IgG to mucosal surfaces. IgG is the predominant antibody produced during the secondary immune response to antigen. Within the IgG class are four major subclasses, designated IgG1, IgG2, IgG3, and IgG4. All IgG subclasses have a similar molecular mass except for IgG3, which has a much longer hinge region than any other IgG subclass. The IgG3 hinge region is approximately four times as long as the IgG1 hinge, containing up to 62 amino acids (including 21 prolines and 11 cysteines), forming a polyproline helix with limited flexibility. Because of this, IgG3 myeloma protein may aggregate spontaneously to produce a hyperviscosity syndrome. Whereas IgG1 and IgG3 proteins activate complement via the classic pathway, IgG2 molecules fix complement poorly and IgG4 proteins not at all. Antibody responses to soluble protein antigens and membrane proteins primarily induce IgG1, but also lower levels of the other subclasses, mostly IgG3 and IgG4. On the other hand, IgG antibody responses to bacterial capsular polysaccharide antigens typically are restricted to IgG2; IgG2 deficiency can result in the virtual absence of IgG anticarbohydrate antibodies. Viral infections generally induce IgG antibodies of the IgG1 and IgG3 subclasses, with IgG3 antibodies appearing first in the course of the infection. As such, IgG4 has been called a "blocking antibody" in the context of allergy, where it may compete with IgE for allergen binding. Physical Properties of Human Immunoglobulins IgG Heavy-chain class Heavy chain subclass No. Biologic Properties of Human Immunoglobulins IgG Percent of body pool in intravascular space Percent of intravascular pool catabolized per day Normal synthetic rate (mg/kg per day) Serum half-life (days) Placental transfer Cytophilic for mast cells and basophils Binding to macrophages and other phagocytes Reactivity with staphylococcal protein A Antibody-dependent cell-mediated cytotoxicity Complement fixation Classic pathway Alternative pathway Yes No No Yes Yes No No No No No 45 6. It is estimated that a normal 70-kg adult secretes approximately 2 g of IgA per day. The most abundant subclass is IgA1, which constitutes approximately 85 percent of the total IgA in plasma. The secreted IgA can bind to a poly-Ig receptor, which is an integral membrane glycoprotein expressed on the basal membrane of mucosal cells. Here the poly-Ig receptor is proteolytically cleaved, releasing the extracellular domain, which remains complexed with the secreted IgA as a 70-kDa secretory protein that can protect the secreted IgA molecule from proteolytic digestion by enzymes in the intestinal lumen. This modified form of IgA, comprised of an IgA dimer bound to the J chain and secretory protein, is the principal antibody in saliva, tears, colostrum, and the fluids of the gastrointestinal, respiratory, and urinary tracts. IgA can direct various effector functions by cells that bear specific Fc receptors for IgA (FcR). Another IgA receptor specific for the secretory protein can elicit powerful effector responses from eosinophils. Their main function may be to prevent foreign substances from adhering to mucosal surfaces and entering the blood. Defective glycosylation of IgA1 can lead to the most common form glomerulonephritis, namely Berger disease or IgA nephropathy. This is an autoimmune disorder in which neoepitopes caused by defective galactosylation of O-linked glycans in the hinge region of human IgA1 are recognized by antiglycan IgG or IgA1 antibodies. IgM molecules classically are termed macroglobulins because of their large molecular weight. IgM macroglobulins do not penetrate easily into extravascular spaces or readily cross the placenta. Compared to monomeric IgG antibodies, pentavalent IgM antibodies fix complement more efficiently. A single IgM molecule on the surface of a red blood cell can initiate complement-mediated hemolysis. Schematic of membrane IgM and its associated acces- IgD IgD is a trace serum protein that composes less than 1 percent of plasma immunoglobulins. IgD is expressed on most peripheral B cells, as is IgM, where it may function as a B-cell membrane receptor for antigen that facilitates recruitment of B cells into specific antigen-driven responses. They do not penetrate extravascular spaces efficiently, cross the placental barrier, or fix complement via the classic pathway. This might explain why mice made deficient in IgD have fewer B cells, delayed affinity maturation, and weaker production of immunoglobulin isotypes, such as IgE, which are highly dependent on such cytokines. IgE has been called reaginic antibody to denote its association with immediate hypersensitivity. In patients with parasitic infestation and in some children with atopic diseases, plasma IgE levels may rise to 5 to 20 times normal. The IgE molecule consists of a four-chain basic unit plus 12 percent carbohydrate. Monomeric IgE binds via the Fc region to high-affinity receptors on the surface membranes of basophils and mast cells. IgM has 10 binding sites for antigen, each composed of a heavy-chain variable region (H-chain V region) and a light-chain variable region (L-chain V region). These substances act on adjacent cells and may regulate the metabolism of the connective tissue extracellular matrix. These lipid mediators and biogenic amines may produce the rapid components of immediate hypersensitivity, such as vascular leakage, vasodilation, and bronchoconstriction. The released cytokines, on the other hand, are responsible for the late phase of the immediate hypersensitivity response. Instead, the immediate hypersensitivity response may represent a pathologic systemic exaggeration of a local physiologic process that ordinarily contributes to the inflammatory response to invading organisms. Such motifs are found in the cytoplasmic domains of several immune system signaling molecules, including those of the T-cell receptor complex (Chap. B cells can become activated following ligation of their surface immunoglobulin receptors by antigen, which typically is presented on the surface of dendritic cells or macrophages. The immunoglobulin heavy-chain gene complex is located at band q32 of the long arm of chromosome 14. The constant-region elements of the heavy-chain gene complex are proximal to variable-region segments on chromosome 14, whereas the constant-region segments of the two light chains are in the opposite orientation, telomeric to the variable-region genes. The heavy-chain genes encoding the constant regions are represented by blue boxes. Similar recognition sequences flank the elements that rearrange to form the T-cell antigen receptor (Chap. If the breakpoints of these chromosomal lesions lie near potential oncogenes or tumor-suppressor genes, they can lead to cellular transformation and lymphoid tumors. However, only one joining event is needed to generate a light-chain gene, whereas two are needed to generate a complete heavy-chain gene. The hairpinned termini of gene segments that give rise to the coding joint each is subsequently cleaved at random sites by an exonuclease. The opened hairpin ends can be modified further by nucleases that can remove a self-complementary overhang or cut further into the original coding sequence. Such processes contribute to immunoglobulin diversity and are the principal mechanism responsible for somatic diversification of the T-cell repertoire (see Chap. It is therefore crucial to limit V(D)J recombination activity to cells within G1 phase; this restriction appears to be controlled by Rag-2 degradation. The specificity of the humoral immune response depends upon antigenic selection of unique clones of B cells, each clone expressing a homogeneous set of immunoglobulin receptors.

Buy 10mg omeprazole mastercard
An independent data monitoring committee stopped the study after a median followup of 1 gastritis diet ������ proven 40 mg omeprazole. The independent data monitoring committee mandated that all patients on the experimental arm be crossed over to receive involved nodal radiotherapy. An analysis of the data according to the treatment actually received revealed a 3-year progression-free survival of 97 percent in the combined modality group compared to 90. Encouraging data from single institutions in adults and children were not borne out in randomized trials, some of which were criticized as underpowered. Furthermore, chemotherapy regimens have evolved over the time span of these studies. Of note, all patients in partial remission received 40 Gy on this study and their outcome was not different from complete remission patients. However, the role of this potent treatment in patients with an early incomplete response or following a brief course of chemotherapy is likely to be more significant. Initial studies were conducted using single-agent rituximab in relapsed and refractory cases and demonstrated response rates of 94 to 100 percent with median durations of remission of 33 to 60 months, with longer remissions observed when maintenance rituximab was used. Improvements in management have resulted in cure for a large majority of patients younger than age 65 years. Survival expectations at 10 years for patients diagnosed from 2006 to 2010 exceed 90 percent for patients to age 44 years, 80 percent for patients to age 54 years, and 70 percent for patients to age 64 years. However, the late effects of treatment for Hodgkin lymphoma remain a concern for cured patients, and a small subset of patients has refractory disease. Consolidation radiotherapy is often employed to sites of pretransplantation bulk disease. The superiority of any single transplant conditioning regimen has not been definitively established; however, the use of high-dose sequential therapy coupled with tandem autologous transplantation is being tested in randomized trials. Allogeneic transplantation in multiply recurrent Hodgkin lymphoma has been limited by significant transplant-related mortality, although long-term disease control has been observed in a small subset together with anecdotal evidence of a graft-versus-Hodgkin antitumor effect. Massive mediastinal disease and constitutional symptoms have been consistently identified as independent predictors of relapse, whereas only older age was predictive of inferior survival. It is important to be aware of the variable eligibility criteria when interpreting the literature in early stage Hodgkin lymphoma and to note that these clinical variables are currently used to group patients for clinical investigations. Only 7 percent of patients were in the worst prognostic group (five to seven factors) and the freedom from progression in this subset was 42 percent at 5 years. Consensus with regard to prognostic factors promotes uniformity in clinical trial design and provides a rationale for Chapter 97: Hodgkin Lymphoma 1617 alternate approaches in high-risk subsets. Fortunately, newer treatment approaches have reduced the proportion of patients in this category. Clinical prognostic factors are surrogates for the underlying cellular and molecular biology of Hodgkin lymphoma. Increased numbers of T-regulatory cells have correlated with favorable outcomes and decreased numbers of markers for cytotoxic T cells have correlated with adverse outcomes in several series. Although the acute complications of chemotherapy and radiotherapy may be troublesome, they are relatively easily managed. Late-treatment effects in the form of sterility, second malignancy, and cardiopulmonary disease are more serious and are known to contribute to shortened longevity for cured patients. As treatment has evolved, the risks of radiation-related complications has lessened but long latency periods and uncertainty regarding associations with lower doses make it difficult to predict individual risks. Recognition and understanding of these problems helps to shape primary treatment choice and facilitate optimal followup for survivors. A large international study observed a significant reduction in excess absolute risk after 1984, presumably as a consequence of change in primary therapy. Patients who have received second-line therapies and autologous transplantation are at highest risk for myelodysplasia and secondary leukemia. There is an increased relative risk of non-Hodgkin lymphomas after treatment for Hodgkin lymphoma. It is not clear how non-Hodgkin lymphomas relate to treatment-related immunodeficiency, predisposition to B-cell malignancy, or a shared common cell of origin. Marginal zone lymphomas with identical B-cell receptor genes also have been identified. The latency for developing second cancers is an important consideration, as these typically develop after at least 10 years and continue to pose excess risk for as long as 30 years after treatment. Cofactors are important for defining the risks of second breast cancer, which are highest for women younger than age 30 years when irradiated and for those who continue to have normal menses. Tobacco exposure has a multiplying effect and alkylating agent exposure also contributes to risk. Among patients with chest irradiation, a tobacco history, and alkylating chemotherapy, the lung cancer risk was 49-fold higher than in patients who had none of these exposures. An increased risk of death from coronary artery disease and acute myocardial infarction has been identified in adults and children. As risk is associated with the dose and volume of radiotherapy and the latency is 5 to 10 or more years, the hazards associated with current lower dose and smaller fields remain to be assessed. Noncoronary vascular complications have been reported after neck irradiation, with associations to dose greater than 36 Gy and cofactors of hypertension, diabetes and hypercholesterolemia. Rarely, hyperthyroidism, Graves ophthalmopathy or thyroid neoplasms occur after neck radiotherapy. Although prospective assessment of pulmonary function demonstrates reduction of lung volumes following mantle radiotherapy, recovery is seen in 12 to 24 months and symptomatic radiation pneumonitis is unusual. Current therapy programs use low-dose or no radiotherapy for all stages of disease. Overwhelming sepsis is a rare event in patients who have been splenectomized and treated for Hodgkin lymphoma, particularly children. However, it must be recognized that neither vaccines nor antibiotic prophylaxis may provide adequate protection. Fatigue is commonly reported in Hodgkin lymphoma survivors and has been related to pulmonary function and peak oxygen uptake. Patient education is essential to promote healthy behaviors to reduce modifiable risk factors. In addition, early detection and prevention strategies for second cancers and cardiac disease should be considered in high-risk patients. However, the choice and efficacy of diagnostic testing, and their optimal timing and frequency, require further study. It continues to be important to follow long-term survivors, and the contribution of genetic and environmental factors is an important ongoing area of inquiry. Fox H: Remarks on microscopical preparations made from some of the original tissue described by Thomas Hodgkin, 1832. Hemminki K, Li X, Czene K: Familial risk of cancer: Data for clinical counseling and cancer genetics. Kuppers R, Rajewsky K, Zhao M, et al: Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Kuppers R, Roers A, Kanzler H: Molecular single cell studies of normal and transformed lymphocytes. Schwering I, Brauninger A, Klein U, et al: Loss of the B-lineage-specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Re D, Muschen M, Ahmadi T, et al: Oct-2 and Bob-1 deficiency in Hodgkin and Reed Sternberg cells. Ushmorov A, Leithauser F, Sakk O, et al: Epigenetic processes play a major role in B-cell-specific gene silencing in classical Hodgkin lymphoma. Cobaleda C, Schebesta A, Delogu A, et al: Pax5: the guardian of B cell identity and function. Emmerich F, Theurich S, Hummel M, et al: Inactivating I kappa B epsilon mutations in Hodgkin/Reed-Sternberg cells. Fiumara P, Snell V, Li Y, et al: Functional expression of receptor activator of nuclear factor kappaB in Hodgkin disease cell lines.
