

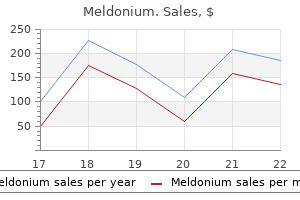
Order 500 mg meldonium otc
Two-thirds were alive at a mean follow-up of 23 months medications listed alphabetically order meldonium with paypal, with a 3-month all-cause mortality of 14%. However, the Boston group has demonstrated an improved quality of life for patients who respond. Nine cardiac transplants were reported from Boston, eight of whom subsequently received a stem cell transplant. At a median follow-up of 56 months, five of seven patients were alive without recurrent amyloid, comparable to patients who received heart transplants for nonamyloid heart disease. At Mayo Clinic, the median survival of patients who have received cardiac transplants for amyloid is approximately 50%, which is somewhat inferior to patients who receive hearts for cardiomyopathy without a systemic disorder. Eleven patients underwent heart transplant followed by autologous peripheral blood stem cell transplant. The median survival was 76 months from heart transplant and 57 months from stem cell transplant. Organ transplant is a viable option for patients in whom complete suppression of light chain production can be achieved. Because of the shortage of organs, critical decisions regarding allocation must be made. Organ transplant has also been applied to familial amyloid polyneuropathy amyloidosis, usually combined with liver transplant. We have performed three cardiac transplants for familial amyloidosis, heart transplant for senile amyloid in 2 patients, heart-liver transplant for familial amyloid in 18 patients, and heart-liver-kidney transplant for familial amyloid in 4 patients. Any patient with unexplained fatigue or restrictive cardiomyopathy or heart failure with preserved ejection fraction should be screened for amyloidosis. Patients who fulfill criteria for chronic inflammatory demyelinating peripheral neuropathy, unexplained hepatomegaly, or atypical multiple myeloma should all be considered for the possibility of amyloidosis. If an immunoglobulin light-chain abnormality is found, it would be appropriate to do biopsies of the bone marrow and subcutaneous fat to stain for Congo red. Only if the index of suspicion is high should further biopsies be performed if the results of bone marrow and fat biopsies are both negative. The gold standard is laser capture mass spectroscopy in an effort to ensure that all chemotherapy-treated amyloid is of immunoglobulin light chain origin. We believe that stem cell transplant is the preferred technique for patients in whom it can be performed safely, but this should not be more than 20% of patients. For nontransplant candidates, melphalan-dexamethasone, melphalan-dexamethasonebortezomib, and cyclophosphamide-bortezomib-dexamethasone are all legitimate options for induction therapy. For patients without Amyloidosis suspected lmmunofixation of serum and urine free light-chain assay Positive Obtain fat and bone marrow specimens for Congo red staining Negative Light-chain amyloidosis unlikely Positive Is amyloid localized (not systemic), such as bladder or larynx Negative Likelihood of amyloidosis 17% Organ biopsy only if a high index of suspicion Yes Refer for local therapy No Could systemic amyloid be non-light chain Mahmood S, Palladini G, Sanchorawala V, et al: Update on treatment of light chain amyloidosis. Shah G, Kaul E, Fallo S, et al: Bortezomib subcutaneous injection in combination regimens for myeloma or systemic light-chain amyloidosis: a retrospective chart review of response rates and toxicity in newly diagnosed patients. Kontoyiannis Advances in the supportive care and treatment of hematologic malignancies have markedly improved the life expectancy of afflicted patients, but this progress is increasingly at the expense of developing a wider range of infectious complications often caused by drugresistant organisms. This article reviews specific hematologic conditions for their unique host defense defects and associated infections (Table 89. Acute Leukemias In patients with acute leukemias, a major cause of morbidity is infection due to drug-associated mucositis and therapy-induced neutropenia. Most infections occurring during neutropenia are bacterial, but patients with prolonged neutropenia are at additional risk for development of yeast and mold infections. Several cytotoxic agents, notably methotrexate, cyclophosphamide, 6-mercaptopurine, and azathioprine, impair cell-mediated immunity. The use of monoclonal antibody therapy for hematologic disorders results in dysfunction of particular aspects of the immune system. Exogenous administration of glucocorticoids leads to increased susceptibility to infection. The degree of immunosuppression and the relative risk for infection depend on the dose and duration of use. The major effect of steroids on granulocyte function is a decrease in chemotactic activity. This accounts, in part, for the clinical observation that the signs and symptoms of severe infections may be masked or greatly reduced in patients receiving steroids. Steroids may enhance susceptibility to infection by means of negative effects on glucose homeostasis, wound healing, skin fragility, monocyte and lymphocyte function, production of cytokines, and humoral immune responses. Radiation therapy has been associated with granulocyte dysfunction and delayed wound healing. Host defense defects with tyrosine kinase inhibitors, such as imatinib or dasatinib, have not been well defined. Lymphomas Hodgkin and non-Hodgkin lymphoma are commonly associated with impaired cell-mediated immunity. The degree of immune impairment may correlate with the extent of disease and often is compounded by administration of immunosuppressive therapy. Splenectomy-related infections occur with sepsis caused by encapsulated bacterial organisms at a median of 22 months but sometimes many years after surgery. Neutrophils from patients with myelodysplastic syndrome have deficiencies in myeloperoxidase, elastase, and integrins. More than half of patients with myelodysplastic syndrome die within 3 years of diagnosis from infections, bleeding complications, or progression to acute leukemia. Chronic neutropenia is the main cause of recurrent bacterial and fungal infections among patients. Treatment of the underlying hematologic disease is required to stop recurrent infections and cure some chronic infections. Cell-mediated immunity is not impaired by the disease but is compromised by corticosteroids or cytotoxic therapy. Paroxysmal Nocturnal Hemoglobinuria Patients with paroxysmal nocturnal hemoglobinuria are at some increased risk for bacterial infection due to a deficiency of decayaccelerating factor on the membrane of neutrophils. Modest and progressive granulocytopenia, progression to aplasia or leukemia may compound these risks. Vaccination against meningococcus is required before treatment with the terminal complement inhibitor eculizumab. Uncommon Malignancies Patients with hairy cell leukemia develop mycobacterial disease relatively often, especially infection with atypical mycobacteria. Reversal of host cellular immune defects with effective therapy can lead to rapid clinical response with eradication of mycobacterial infection. Granulocytic Phagocyte Disorders the clinical approach to infections in patients with granulocytic phagocyte disorders is specific to each of these disorders and is beyond the scope of this chapter. Chronic granulomatous disease is a heterogeneous group of disorders resulting from defective or malfunctioning oxidative metabolism capacity of phagocytes. Recurrent infections with bacteria and fungi are common and occasionally life threatening, despite optimal antimicrobial therapy. Infections with Staphylococcus species and Aspergillus species can be particularly aggressive. Deficiency of this enzyme limits glucose metabolism through the hexose monophosphate shunt, resulting in an abnormal respiratory burst in neutrophils. Defects in cellmediated immunity have been described in patients with thalassemia. Patients with sickle cell disease have an increased susceptibility to bacterial infections. Splenic involution results in depressed synthesis of alternate pathway factor(s) of complement and decreased phagocytic clearance of bacteria. Phagocytosis of Streptococcus pneumoniae is abnormal, in part because of an inability to use the alternate pathway for C3 fixation as a means of opsonization. An increased risk for Salmonella infection appears to be unique to the sickle cell population. Suppurative arthritis can occur after repeated episodes of hemarthrosis among patients with sickle cell disease.
Diseases
- Myoglobinuria recurrent
- Chromosome 6, monosomy 6q2
- McArdle disease
- Macrodactyly of the foot
- Alves Dos Santos Castello syndrome
- Pulmonary blastoma
Discount 250 mg meldonium
Membrane proteins may be crosslinked symptoms tracker purchase meldonium 250mg mastercard, with binding of denatured, oxidized hemoglobin to the membrane cytoskeleton, which may increase splenic macrophage recognition. The oxidative lesions can be severe enough to cause intravascular destruction as well, producing hemoglobinemia and hemoglobinuria. The smear may show bite cells, which look as if a macrophage had taken a bite, removing a Heinz body-containing segment of membrane. Severe hemolysis may produce the kind of circulating ghost or hemighost called a blister cell or bite cell. Profound cyanosis with methemoglobinemia can occur within hours, with levels of 120% or higher. Toxic ingestion or inhalation of nitrites may occur in suicide attempts from industrial exposures; via diets high in pickled or smoked foods; through intentional recreational use; or in infants from formulas prepared using well water high in nitrates, which are reduced to nitrites in the infant gut. Benzocaine topical anesthesia in the form of a spray or cream can cause severe methemoglobinemia, with cyanosis and dyspnea requiring methylene blue treatment. Pyridium (phenazopyridine) can cause oxidative hemolysis even in the absence of renal disease. It has been recognized for more than 130 years that therapy with dapsone causes oxidative hemolysis. In the past, dapsone was used primarily to treat leprosy and dermatitis herpetiformis, and was not often encountered as a cause of oxidative hemolysis. Zuber et al30 reported eight patients with chronic myeloid leukemia who developed thrombotic microangiopathy confirmed by renal biopsy. Seven of these patients had identifiable hemolysis, and three had thrombocytopenia. However, preparations of IgG contain anti-A and anti-B antibodies, and rarely cause an alloimmune hemolytic anemia, as described in two young women undergoing treatment for idiopathic thrombocytopenic purpura. If this situation occurs and more intravenous IgG is needed, performing a minor cross-match and choosing a preparation of intravenous IgG that gives no reaction is recommended. In addition to isoantibody production, anemia has been reported with intravenous IgG because of immune complexmediated complement activation. For hemolytic anemia with large granular lymphocyte leukemia, see box on Hemolytic Anemia in Chronic Large Granular Lymphocytic Leukemia. Looareesuwan S, Ho M, Wattanagoon Y, et al: Dynamic alteration in splenic function during acute falciparum malaria. Veldhuis W, Janssen M, Kortlandt W, et al: Coombs-negative severe haemolytic anaemia in an immunocompetent adult following cytomegalovirus infection. Zuber J, Martinez F, Droz D, et al: Alpha-interferon-associated thrombotic microangiopathy: a clinicopathologic study of 8 patients and review of the literature. There have been no formal studies on therapy for large granular lymphocytic leukemia-related hemolysis, although immunosuppressive therapy with prednisone or methotrexate has been reported as being partially or wholly successful. The clinical significance of leukocytosis or leukopenia varies from none at all to being an early clue to a life-threatening process, whether a primary hematologic or secondary reactive process. This article considers disorders faced by adult practitioners in hospital and outpatient clinics where the predominant hematologic abnormality is neutrophilic leukocytosis, neutropenia, monocytosis, or monocytopenia; other chapters consider lymphocytosis, lymphopenia, eosinophilia, pancytopenia, and hematologic neoplasms. The normal range for leukocyte count in most laboratories is from about 4500/mm3 to 11,000/mm3. Neutrophils (and band forms) comprise the majority of circulating leukocytes (1800 mm3 to 7700/ mm3); monocytes are about 4% of cells (mean absolute count: 300/ mm3). The physician must always think in terms of absolute counts of leukocyte subpopulations (total leukocyte count multiplied by the differential percentage). When approaching a patient with abnormal leukocyte number, several factors impact heavily on the differential diagnosis and the vigor with which diagnosis and therapy should be pursued. Diagnostic considerations are vastly different when the abnormality first manifests in the hospital versus in the outpatient clinic. Also crucial is the degree of the abnormality, providing guidance to its likely cause and consequence. For example, agranulocytosis is a life-threatening disorder in which neutrophils are at or near zero, has a limited spectrum of underlying causes (drug reactions being paramount), and demands immediate interventions. Duration has major implications; determining the onset of changes and whether they are stable or progressive informs as to etiology and significance. Whether the abnormality is symptomatic-for example, whether a neutropenic or monocytopenic patient has or has had infectious complications-bears on likely etiologies and need for therapy. If there are known or suspected comorbid conditions, such as autoimmune or inflammatory disorders, this can crystallize the approach; occasionally, the leukocyte abnormality may be the first sign of a previously unrecognized disorder or may provide important confirmation. Beyond history and physical examination, the peripheral blood smear is key to establish the direction of further evaluation. In the emergency department, leukocytosis is often equated with significant bacterial infection or is at least a sign of illness severe enough to warrant hospital admission rather than outpatient management. Patients with leukoerythroblastosis do not necessarily have leukocytosis, but they usually do. Teardrop poikilocytes and elliptocytes on blood smear would strengthen concerns for myelophthisis. Left-shifted neutrophils refer to relative immaturity of circulating cells, often manifest as an increased percentage of band neutrophils. Marked left-shift includes less mature precursor forms, myelocytes and metamyelocytes. Left-shift is nonspecific and may occur with infection or any cause of marked neutrophilia. Detailed directed history and physical examination are indispensable to the evaluation of neutrophilia (Table 48. Fever and chills suggest infection (or inflammation), mandating a search for more specific symptoms that could pinpoint the focus. Examples include a sore throat, pharyngeal erythema, and exudate in pharyngitis; productive cough and abnormal lung auscultation in pneumonia; and dysuria and flank tenderness in urinary tract infection. Recent vigorous exercise, emotional stress, burns, shock, or trauma can increase circulating neutrophils because of catecholamine-induced demargination. Often neglected are attempts to delineate the time course of the leukocyte abnormality by seeking prior medical contacts and blood count results at the time. On the physical examination, care should be directed to lymph node palpation because this can be an important clue for infection or malignancy. Palpable splenomegaly may not only direct the evaluation toward hematologic disorders but can be a cardinal sign of a variety of infectious and inflammatory disorders. Blood smear should always be a part of initial evaluation when there are abnormalities of blood counts. Infection To protect against the ever-present threats to health and longevity, evolution has armed us with reactant cytokine cascades designed to increase the number of phagocytes and dispatch them to threatened locales. Neutrophilia is also a frequent response to other types of infections, such as fungal, parasitic, mycobacterial, and sometimes viral. Changes in neutrophil morphology may be useful in predicting whether bacterial or other infection underlie a neutrophilic response. The authors have confirmed some published reports that prominent neutrophil vacuolization is highly specific and moderately sensitive for serious bacterial infection, as are prominent Dohle bodies (in the absence of a primary hematologic disorder). The authors blindly scored blood smears from 50 patients with serious bacterial infection (half bacteremic), 25 with influenza, 25 with noninfectious fever, and 25 control smears. Toxic granulation of neutrophils, touted as a sign of bacterial infection, was found useless in distinguishing infections from other febrile illnesses. In patients with leukemoid reaction, a disproportionate number have infection with Clostridium difficile, an organism that elicits a vigorous neutrophil response. Leukemoid reactions may be associated with solid tumors, sometimes due to paraneoplastic production of colony-stimulating factor. It is important to quickly differentiate a reactive neutrophilic leukocytosis from a clonal leukemic proliferation. Most patients with leukemoid reaction are encountered very ill in the hospital with obvious underlying illnesses. Physiologic stresses, including exercise and emotional stress, lead to endogenous catecholamine and glucocorticoid release in addition to inflammatory cytokines.
Purchase 250mg meldonium fast delivery
Formation of methemoglobin and it physiologic (open space) and therapeutic reduction (shaded space) symptoms your dog is sick buy meldonium in united states online. Transaldolase deficiency presents in the neonatal period and clinical manifestations include dysmorphic features, hepatosplenomegaly, cirrhosis, cardiac and renal abnormalities, skin manifestations, and thrombocytopenia, although the phenotype is highly variable. Methemoglobin is a derivative of hemoglobin in which the ferrous (Fe2+) irons are oxidized to the ferric (Fe3+) state. Methemoglobin is formed spontaneously at a slow rate by the autoxidation of hemoglobin. Methemoglobin may also be formed from the oxidation of hemoglobin in other reactions with endogenous and exogenous compounds. The ferric hemes of methemoglobin are unable to bind oxygen and, additionally, if a ferriheme subunit is part of a hemoglobin tetramer, the oxygen affinity of the accompanying ferrous hemes in the hemoglobin tetramer is increased. As a result, the oxygen dissociation curve is left-shifted and oxygen delivery is impaired. Methemoglobin is formed continuously, but reducing mechanisms keep the methemoglobin level at about 1% of the total hemoglobin. The consequent left shift of the oxygen dissociation curve increases hemoglobin affinity for oxygen, thus resulting in decreased delivery of oxygen into the peripheral tissues and compensatory polycythemia. Since these electron acceptors are not physiologic, this pathway is only of importance as it is the mechanism by which methylene blue treats acute toxic methemoglobinemia. Epidemiology Most cases of methemoglobinemia are acquired, resulting from increased methemoglobin formation by various exogenous agents. Acute or toxic methemoglobinemia may occur in the setting of overdose or poisoning, but also at standard doses of drugs. Acute methemoglobinemia occurs equally between males and females and over a wide range of ages; however, infants are more susceptible because their erythrocyte b5R activity is normally 50% to 60% of adult activity. Other causes of hereditary methemoglobinemia are the autosomal dominant inheritance of an abnormal hemoglobin in hemoglobin M disease (see Chapter 43) and, very rarely, deficiency of cytochrome b5. Death typically ensues at methemoglobin levels above 70% but can occur at lower levels. Individuals with type I b5R deficiency, which is limited to erythrocytes, have methemoglobin concentrations of 10% to 35% and appear cyanotic but are usually asymptomatic, even with levels up to 40%. Other neurologic symptoms may be present, including microcephaly, opisthotonus, athetoid movements, strabismus, seizures, and spastic quadriparesis. LaboratoryManifestations the laboratory diagnosis of methemoglobinemia is based on analysis of its absorption spectra. A fresh specimen should always be obtained because methemoglobin levels tend to increase with storage. Traditional pulse oximetry is unreliable in the presence of methemoglobinemia because of its light absorbance properties; however, co-oximetry can determine the methemoglobin fraction along with all other substances with the optical density at 630 nm. Methemoglobin detected by co-oximeter should be confirmed by the specific Evelyn-Malloy method if available. This method involves direct spectrophotometric analysis and should be used when methemoglobinemia is suspected. In the Evelyn-Malloy method, blood is lysed in a slightly acid buffer and the optical density is measured at 630 nm before and after adding a small amount of neutralized cyanide Absorption of methemoglobin at this wavelength disappears when it is converted to cyanmethemoglobin. This method remains the most accurate technique for the estimation of methemoglobin concentration. Distinguishing the hereditary forms of congenital methemoglobinemia requires interpretation of family pedigrees as well as biochemical analyses. Cyanosis in successive generations suggests autosomal dominant hemoglobin (Hb) M disease, whereas normal parents but possibly affected siblings implies autosomal recessive b5R deficiency. Because the enzyme defect is found in fibroblasts, analysis of b5R activity in cultured amniotic cells for prenatal diagnosis is possible. Pathobiology Acute Methemoglobinemia Many drugs and toxins have been implicated in acute methemoglobinemia. More common culprits include dapsone, local anesthetics (benzocaine, lidocaine, prilocaine), and derivatives of the anesthetic phenacetin. Exposure to nitrates and nitrites, widely used as food preservatives and found in well water can also cause methemoglobinemia. Nitrates do not oxidize hemoglobin directly but are converted to nitrites by intestinal bacteria. Infants less than 6 months of age may have increased susceptibility to methemoglobinemia at least in part because of their lower b5R activity. Homemade baby food purees of high-nitrate-containing vegetables, well water contaminated by nitrites and diarrheal illness may all cause acute toxic methemoglobinemia in infants. B5R Deficiency In erythrocytes, b5R transfers electrons to methemoglobin to reduce it to hemoglobin. In other cells, b5R transfers electrons from cytochrome b5 to stearyl-CoA in the endoplasmic reticulum, a reaction that has an important role in cholesterol biosynthesis, fatty acid elongation and desaturation, and drug metabolism. The more common type I b5R deficiency is usually caused by missense mutations leading to decreased stability of the enzyme. Other pigments, including methylene blue, may also produce false positive results when methemoglobin is measured by co-oximetry. ClinicalManifestations Methemoglobinemia causes clinically discernible cyanosis when the absolute level of methemoglobin exceeds 1. Methemoglobinemia should be clinically suspected when "cyanosis" occurs in the presence of a normal PaO2. Symptoms develop secondary to impaired tissue oxygenation and the onset may be abrupt. At higher levels, respiratory depression, altered consciousness, shock, seizures, and death may occur. As methemoglobin levels rise above 20% to 30%, Prognosis Acute methemoglobinemia generally resolves promptly with treatment providing the offending cause is discontinued. Therapy Offending agents in cases of acquired methemoglobinemia should be discontinued. However, if the patient is symptomatic or if methemoglobin levels are greater than 20%, specific therapy is indicated. Leukomethylene blue, in turn, nonenzymatically reduces methemoglobin to hemoglobin. In these patients, methylene blue would not only fail to give the desired effect on methemoglobin levels but might compound the situation by inducing an acute hemolytic episode. The cyanosis in hereditary b5R deficiency is of cosmetic significance only but can be treated with methylene blue or ascorbic acid, both of which facilitate the reduction of methemoglobin through alternate pathways. Beutler E: Red cell metabolism: a manual of biochemical methods, ed 3, 1984, Grune & Stratton. A summary of current understanding and management of clinical and metabolic features of pyruvate kinase deficiency. Summary of red cell enzyme defects in descending order of their clinical importance. This review focuses on the impact of energy metabolism of erythrocyte and its pathophysiology. Viprakasit V, Ekwattanakit S, Riolueang S, et al: Mutations in Kruppellike factor 1 cause transfusion-dependent hemolytic anemia and persistence of embryonic globin gene expression. Review of red cell enzymes with extensive bibliography of original and recent articles. In patients with severely dysfunctional spectrin mutations, the weakened spectrin dimer-dimer self-association disrupts the skeletal lattice, leading to a marked skeletal instability and cell fragments. It is speculated that elliptocytes are permanently deformed cells because the weakened horizontal interactions facilitate a shear stress-induced rearrangement of skeletal proteins, precluding recovery of the normal biconcave shape. Acanthocytosis,Stomatocytosis,andtheBilayer CoupleHypothesis the mechanism of acanthocytosis and stomatocytosis associated with defects of membrane proteins is much less clear. Most forms of acanthocytosis are associated with either acquired or inherited abnormalities of membrane lipids. In rare cases with acanthocytosis, membrane protein abnormalities have been detected, but the associated mechanisms leading to acanthocyte formation are unknown. These abnormalities occur in the McLeod phenotype, the choreaacanthocytosis syndrome, and other rare disorders.
Best purchase meldonium
In the Mayo Clinic experience rust treatment generic meldonium 500mg with mastercard,25 cladribine was administered for 5 days at a dose of 5 mg/m2/day (or 0. The median response duration was 11 months, and inferior outcomes were predicted by leukocytosis, monocytosis, and circulating immature myeloid cells, the latter remaining significant in a multivariate analysis. The overall response rate was 72%, split between 92% and 50% for indolent and advanced disease, respectively. Lymphopenia (82%), neutropenia (47%), and opportunistic infections (13%) were the most common grade 3/4 adverse events. Subjects were randomized to oral masitinib (6 mg/kg daily in two daily doses) or placebo. The primary end-point of the study was based on a 75% or more improvement in one or more symptom categories: pruritis, flushing, depression, or fatigue. At week 24, there was an 18% decrease in the serum tryptase level in the masitinib arm versus an increase of 2. Also, urticaria pigmentosa lesions on masitinib therapy decreased by an average body surface area of 12. Masitinib-associated clinical benefits were generally sustained during a two-year extension period. Although treatment was generally well tolerated, there was an excess incidence of diarrhea, rash, and asthenia in 9%, 6%, and 4% of patients, respectively, in the masitinib group. In addition, 24% of patients in the masitinib arm discontinued therapy because of an adverse event, compared with 10% in the placebo arm. However, the safety and efficacy of this approach has yet to be validated in the context of clinical trials. However, clinical trial data regarding their activity in patients have not yet been published. A clinical trial testing brentuximab has recently been initiated in the United States. The remaining 30% of responses were split between stable disease (21%) and primary refractory disease (9%). In addition, inferior survival was observed in patients undergoing reduced-intensity versus fully myeloablative conditioning. Regulatory health agencies are increasingly focused on these patient measures for drug approval, and validated patient-reported outcomes are critical for stringent adjudication of treatment-related changes in the context of placebo-controlled, double-blind study designs. These original criteria or their modified version have been used to adjudicate responses in clinical trials of new agents. Second, responses in C-findings such as ascites, weight loss, and bone lesions are often difficult to quantify. Third, criteria for baseline red blood cell and platelet transfusion dependence were not codified. Valent P, Akin C, Escribano L, et al: Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Schwaab J, Schnittger S, Sotlar K, et al: Comprehensive mutational profiling in advanced systemic mastocytosis. Pardanani A: Systemic mastocytosis in adults: 2015 update on diagnosis, risk stratification, and management. Barete S, Lortholary O, Damaj G, et al: Long-term efficacy and safety of cladribine (2-CdA) in adult patients with mastocytosis. Valent P, Akin C, Arock M, et al: Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus protocol. Valent P, Escribano L, Broesby-Olsen S, et al: Proposed diagnostic algorithm for patients with suspected mastocytosis: a proposal of the European Competence Network on Mastocytosis. The predominant cell type is a small lymphocyte with clumped chromatin, but a spectrum of nuclear morphology is usually seen. Pseudofollicular growth centers or proliferation centers are present in the majority of cases and contain a spectrum of cells ranging from small lymphocytes to prolymphocytes and paraimmunoblasts. The prolymphocytes and paraimmunoblasts have more dispersed chromatin and more prominent nucleoli usually centrally placed. If needed, immunophenotypic studies can be helpful in this differential diagnosis. The latter two have been used as partial surrogate markers for the mutational status. Treatment with immunosuppressive agents such as fludarabine appears to increase risk. The current approach is based on the integration of morphologic, phenotypic, genetic, and clinical features that allows the identification of distinct disease entities (See box on Principles of the Classification of Lymphomas). Furthermore, it recognizes that any classification system to be viable and applicable should evolve and incorporate new data resulting from emerging technologies in the field of hematopathology such as results from genome-wide large-scale sequencing studies. These studies have led to the identifications of new prognostic and diagnostic categories, and provide insight into therapeutic targets based on a better understanding of molecular mechanisms of transformation. Even in patients with a lymphomatous presentation, careful examination of the blood may reveal a circulating monoclonal B-cell component. However, for most diseases, knowledge of the immunophenotype and molecular genetics/cytogenetics plays an important role in differential diagnosis. A disease-based approach to classification facilitates discovery of molecular pathogenesis the sites of presentation and involvement are important clues to underlying biologic distinctions. Many lymphoma entities display a range in cytologic grade and clinical aggressiveness, making it difficult to stratify lymphomas according to clinical behavior. A number of prognostic factors influence clinical outcome, including stage, international prognostic index, cytologic grade, gene expression profile, secondary genetic events, and the host environment. These early lesions can in some ways be considered equivalent to benign neoplasms in the epithelial system, and require special management approaches. Early lesions appear to lack the secondary and tertiary "hits" seen in lymphoid neoplasms that are clinically significant, and most patients have a very low risk of clinical progression. These have fairly round nuclear contours, condensed nuclear chromatin, and inconspicuous or absent nucleoli. Small lymphocytic lymphoma can transform to large cell lymphoma (C) and occasionally to Hodgkin lymphoma (D). Patients can also develop worsening lymphadenopathy from viral infections such as herpes simplex virus, in which the node typically shows focal necrosis (E). There is a diffuse infiltrate (A) of small lymphocytes that have plasmacytoid features or interspersed plasma cells ((B), bone marrow; (C), lymph node). Evaluation for and by immunohistochemical stains can demonstrate clonality in the plasma cells and plasmacytoid lymphocytes (D). In the latter, the neoplastic mantle zones are expanded and can become confluent leaving "naked" germinal centers (A). They have irregular nuclear contours, especially compared with small lymphocytic lymphoma, and they have dense chromatin. Some cases can develop a "blastoid" transformation (D), although some cases can present as a "blastoid" variant. Such cases are characterized by cells with an intermediate size, a high mitotic rate, and finely dispersed "blastic" chromatin. Sometimes when the "blastoid" cases develop a leukemic phase, they can be difficult to distinguish morphologically from acute lymphoblastic leukemia. In such cases, flow immunophenotyping is needed to resolve the differential diagnosis. The latter refers to a reactive lymph node with Cyclin D1 positive cells limited to an otherwise normal appearing follicle mantle; these cases tend not to progress and should not be labeled as lymphomas. These patients have an indolent clinical course and do not appear to require aggressive chemotherapy.
Aspidosperma quebracho-blanco (Quebracho). Meldonium.
- Dosing considerations for Quebracho.
- What is Quebracho?
- How does Quebracho work?
- Are there safety concerns?
- Asthma, lung disorders, cough, high blood pressure, spasms, fluid retention, menstrual cramps, fever, increasing sex drive, and other conditions.
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96400
Buy meldonium no prescription
These regimens combine drugs with varying mechanisms of action at different doses medicine gustav klimt order meldonium 500 mg on-line, often in complex schedules, which have largely evolved empirically. Because of these challenges and the relative rarity and biologic heterogeneity of the disease, it is recommended that patients are referred to larger academic centers for evaluation and treatment to ensure the best possible survival rates. Effective chemotherapy regimens must be supplemented with adequate supportive care measures to obtain the best patient outcomes. It is also reasonable to consider antifungal and antiviral prophylaxis during periods of neutropenia. Consideration may be given to antibacterial prophylaxis during periods of neutropenia. Fluoroquinolones such as levofloxacin are the preferred agent, although the optimal antibiotic choice should depend on local bacterial resistance patterns. Patients must be provided adequate hydration and prophylaxis or treatment for hyperuricemia with allopurinol for the first week of induction. Rasburicase, a urate oxidase, can be considered for the treatment of hyperuricemia in cases with high proliferation rates. Once a specific regimen has been selected, the goal (and challenge) is for both the medical team and the patient to adhere to the recommended treatments, which are arduous and lengthy in duration. Studies in pediatric patients have shown lower relapse rates but higher toxicity (more sepsis, higher incidence of osteonecrosis) with dexamethasone. Addition of pulsed cyclophosphamide to induction chemotherapy has been studied with conflicting results. Depletion of asparagine results in inhibition of protein synthesis and subsequent apoptotic leukemic cell death. However, this preparation can be immunogenic, leading to hypersensitivity reactions and development of cross-reacting antibodies; this preparation, while still used elsewhere, is no longer available for administration in the United States. After the single dose, asparagine deamination was complete in all patients after 2 hours and in 100%, 81%, and 44% on days 14, 21, and 28, respectively. Of the 85 evaluable patients, those with effective asparagine depletion (defined by enzyme levels >0. Thus, use of pegaspargase is generally safe and effective in adults, and has replaced native L-asparaginase as the agent for asparagine depletion. Patients receiving Erwinia asparaginase are still at risk for known toxicities of asparaginase, including bleeding, clotting, transaminitis, and pancreatitis. The achievement of rapid cytoreduction during this steroid prophase has also been shown to be of prognostic value in several studies. Conventionally, consolidation and maintenance chemotherapy has been the recommendation for this group of patients, given lower potential for toxicities and up to 40% to 60% survival at 5 years with this approach. The specific drugs and the sequence or combinations in which they used are different for each protocol but typically include cyclophosphamide, L-asparaginase (or pegasparaginase), methotrexate, cytarabine, 6-mercaptopurine, vincristine, and doxorubicin. Many of these protocols have evolved empirically with few randomized trials evaluating the impact of any of the modifications that have been developed (see Table 66. They identified the following factors to be independent poor prognostic factors for survival: older age, Ph+ disease, leukocytosis, thrombocytopenia, poor performance status, and hepatomegaly. Histologic section of the brain is illustrated from a patient with acute lymphoblastic leukemia and leukemic meningitis. These patients should receive intrathecal chemotherapy (intrathecal methotrexate alone or together with cytarabine and steroid [triple intrathecal therapy]) twice weekly until clearing of cerebrospinal fluid along with initiation of systemic chemotherapy. Although this has not been demonstrated in adult studies, definitive evidence from pediatric studies indicates that avoidance of cranial irradiation may prevent declines in cognitive function. Although the benefit of maintenance therapy in adult trials has not been demonstrated in randomized trials, it has been shown that omission of maintenance treatment leads to worse clinical outcomes. The study was stopped earlier than planned because remission duration was shorter than in historical control participants who had received maintenance therapy. Thus, it appears that careful adjustment of the doses of the oral agents used during maintenance therapy to achieve optimal yet safe myelosuppression improves treatment outcomes. In a subgroup analysis, the survival benefit of transplant was restricted to patients who were younger than 35 years of age. They used imatinib 400 mg/day on days 1 to 14 of each of the intensive chemotherapy courses, followed by imatinib at a dose of 600 mg daily during the maintenance phase, with monthly vincristine and prednisone, and then imatinib indefinitely. The Japanese have also reported impressive improvements compared to their control participants. Later maintenance therapy with imatinib 600 mg/day with monthly vincristine and prednisone was given. This is the first prospective randomized controlled trial comparing imatinib with chemotherapy and clearly showed the superiority of the imatinib arm. Other potential mechanisms of resistance that have been described include reduced intracellular availability of imatinib and activation of alternative signaling pathways such as the Src-kinase pathways. The study was amended to allow dasatinib 100 mg/day for 14 days during cycle 1, and then 70 mg/day continuously during subsequent cycles. Maintenance chemotherapy consisted of daily dasatinib 100 mg/day with monthly vincristine and prednisone for 2 years followed by dasatinib indefinitely. All patients achieved a complete hematologic response, and there were no induction deaths. At day 85, 58 out of 60 (97%) patients achieved a complete hematologic remission; however, only 11 (18. Thirty five (95%) patients achieved major molecular remission and 26 (70%) complete molecular remission. Relapse occurred in 13% (7 out of 54) of the transplanted patients compared with 90% (18 out of 20) in those who were not transplanted. Imatinib was discontinued early in more than half the patients in both groups, mostly because of gastrointestinal toxicity. High complete molecular remission rates can now be achieved and have been demonstrated to be associated with improved outcomes. Bone marrow biopsy and aspirate features of Burkitt leukemia/lymphoma are illustrated. The bone marrow (or lymph node) will show sheets of highly proliferating intermediate-sized neoplastic cells with a syncytial appearance. The cells are monotonous but have a stippled intermediate chromatin pattern with multiple small nucleoli. Immunophenotypically, the tumor cells express moderate to strong levels of surface immunoglobulin M with light chain restriction, indicating origin of the tumor from a mature B cell. Prompt diagnosis and recognition of this entity is essential because this is now a highly curable leukemia (in the range of 65% to 80% in recent trials); however, failure to institute appropriate therapy at diagnosis for Burkitt lymphoma/leukemia results in emergence of early resistance and dismal outcomes. All current regimens rely on short intensive courses of chemotherapy that incorporate fractionated doses of alkylating agents, high doses of methotrexate and cytarabine, and intensive intrathecal prophylaxis. Recurrence after the first 2 years rarely occurs; therefore maintenance therapy has not been shown to be beneficial and is not recommended. Because of community referral patterns, these patients may be treated by either pediatric or adult oncologists. As such, the treating physician may view a patient in this age group either as an older child or as a younger adult. The oncologist will choose a regimen most appropriate for the population usually seen by that particular physician. A number of comparisons of the clinical outcome of adolescents enrolled on adult and pediatric clinical trials (Table 66. The first of these trials to have been reported highlights many of the interesting questions posed by these comparisons and is reviewed briefly here. The two patient groups were well matched for biologic features, including immunophenotype and cytogenetics. Although the treatment approaches differ among countries, several treatment themes have emerged as being potentially important. Pediatric studies throughout the world use considerably more treatment with nonmyelosuppressive drugs, including glucocorticoids (both dexamethasone and prednisone), vincristine, and L-asparaginase.
Cheap meldonium 250mg free shipping
These patients have distinct molecular phenotypes treatment research institute buy 250 mg meldonium free shipping, with increased expression of chromosome 21 genes in patients with constitutional trisomy 21 as compared with M7 leukemia patients without constitutional trisomy 21. The frequency of these recurrent monosomies are: -7 and -17 in 6%, -18 in 5%, -5 and -21 in 4%, -20 in 3%, -3, -12, and 22 in 2% and loss of chromosomes 2, 4, 9, 13, and 19 in 1%. Each of the autosomes and sex chromosomes can contribute to the numerical changes. The most common trisomies in decreasing order of frequency are gain of chromosome 8, 22, 13, 21, and 11. In the latter situations, +8 does not appear to adversely affect the favorable outcome of patients with t(15;17), inv(16)t(16;16), and t(8;21). By contrast, patients with +8 and a complex karyotype and/or an unfavorable aberration such as del(5q) or -7 usually have a very poor outcome. Isolated +8 has been considered to be associated with either intermediate or unfavorable prognosis. The abnormalities of 17p are often associated with other chromosomal aberrations such as del(5q), -5, -7, but is also an independent poor-risk prognostic factor. These erythromegakaryoblastic leukemias are diagnosed before the age of 5 years and often present with thrombocytopenia and/or myelodysplasia. In contrast, cytogenetic results influence treatment decisions by conferring unfavorable risk assignment on patients with negative broad molecular screening. These methodologies provide complementary genetic information for diagnosis, treatment, and follow-up. Approximately 80% of all patients with a complex karyotype have deletion 5q, followed by deletions 17p and 7q, occurring in approximately 50% of cases. The biologic consequences of these small genomic imbalances are not fully understood. The spectrum of cytogenetic abnormalities in older adults includes a higher percentage of patients with abnormalities involving -5/del(5q), -7/del(7q), and 17p and a lower incidence of translocations associated with a favorable prognosis and treatment outcome. A normal karyotype is observed in 8%, and an abnormal karyotype is detected in 92% of the patient population. Recurrent abnormalities of chromosomes 5, 7, or both account for 70% of all abnormalities observed in therapy-related leukemia. Each of these disorders is characterized by recurrent mutations in specific genes, some of which are shared between several different phenotypes. These tests are performed at diagnosis to establish a benchmark for the percentage of neoplastic cells and are used in follow-up studies to assess the effectiveness of therapy. GeneticTestingforTherapy-RelatedNeoplasms the best genetic test for therapy-related myelodysplastic syndrome or acute myeloid leukemia is a conventional cytogenetic study. The darker gold or blue color indicates those subtypes generally associated with poor prognosis. The application of contemporary genome-wide molecular analysis continues to reveal many additional genetic rearrangements that are not detectable with chromosome studies. In adults the low-risk category includes high hyperdiploidy and del(9p), whereas the high-risk category includes hypodiploidy/near triploidy, t(9;22)(q34;q11), t(4;11)(q21;q23), t(8;14)(q24;q32), and a complex karyotype (five or more chromosomal abnormalities). The reason for this discrepancy may be that adults often have poor-risk chromosomal translocations, such as the Ph chromosome. Approximately 50% of the high hyperdiploid patients harbor other structural abnormalities, such as gains of 1q, del(6q), which do not appear to influence prognosis, with a possible exception of prognostically adverse isochromosome 17q. This translocation is difficult to detect by conventional cytogenetics because the translocated portions of 12p13 and 21q22 have virtually identical G-banding patterns. Prospective analyses have demonstrated that the survival rate in t(12;21)-positive patients is significantly better when compared with cases lacking this abnormality; however, this abnormality in multivariate analysis was not found to be an independent predictor of outcome. Intermediate Favorable/unfavorable Not prognostic Not prognostic Not prognostic Poor Excellent Excellent, intermediate High risk Poor Treated on high risk protocol See Table 56. The most common secondary change, which occurs in approximately 50% of cases with additional abnormalities, is trisomy 21. In these patients, amplification is initiated by a chromothripsis event (a process whereby localized genomic regions are shattered and rearranged in one catastrophic event) that affects both sister chromatids of the Robertsonian chromosome. The end-product is a derivative of chromosome 21 or the rob(15;21)c chromosome with gene dosage optimized for leukemic potential, showing constrained copy number levels over multiple linked genes. Note that the first chromosome 9 has deletion of the short arms, a frequent finding in both adult and pediatric patients with acute lymphoblastic leukemia. Recent studies have indicated that kinase domain mutations are present in 70% of imatinib resistant patients with T315I (37%), E255K (18%) and Y253H (18%) mutations accounting for 75% of these cases. Most frequent are gain of Ph chromosome, monosomy 7, +8, +X, del(9p), and high triploidy. The additional chromosomal abnormalities have no effect on survival of these patients. Other rearrangements involving 12p include deletions, duplications, and translocations and are observed most often as part of a complex karyotype, frequently associated with chromosome 5 and/ or 7 abnormalities. Cytogenetically, two forms of t(1;19) have been identified: 25% of cases have a balanced reciprocal t(1;19), whereas 75% have a rearrangement of unbalanced der(19)t(1;19)(q23;p13. The unbalanced der(19)t(1;19) arises from the initial trisomy of chromosome 1 followed by the t(1;19) translocation, with subsequent loss of the derivative chromosome 1. One-third of patients with t(4;11) have secondary abnormalities; the most frequent are +X, i(7q), abnormalities of 9p, including i(9q), and +8. The outcomes of patients with t(11;19) is generally poor, especially in children younger than 1 year of age. The most frequent additional abnormalities in patients with t(11;19) are +X, +8, and del(6q). Variant translocations t(8;22) (q24;q11) and t(2;8)(p12;q24) are seen in less than 1% of children and adults. This subgroup frequently have a gain of X chromosome, trisomy 21 as an acquired abnormality, and the Ph chromosome/t(9;22) (;q34;q11). Also, in a sharp contrast to the myeloid neoplasms, they almost never occur in infants. Top panel shows a partial G-banded karyotype of chromosomes 8 and 14 with arrows indicating the breakpoints on each chromosome. Greaves found that most common chromosomal translocations and their resultant gene fusions can be documented by molecular analysis of neonatal blood spots or Guthrie cards. The concordance rate of twins who share a monochorionic placenta and develop leukemia is nearly 100%, whereas older twins have a discordance rate of 90%, indicating that additional postnatal leukemic events are needed. The prenatal mutation occurs commonly, exceeding the actual rate of developing leukemia by some 100-fold, indicating a low rate of reentrance or evolution. Moreover, a complex conventional karyotype remains an independent prognostic indicator associated with a poor outcome. Four genomic aberrations, as well as normal findings, are independent predictors of disease progression and survival. Genomic aberrations of prognostic significance include 17p deletion, 11q deletion, trisomy 12, and 13q deletion. Survival of patients with a normal karyotype was 111 months, and the treatment-free interval was 49 months. Two risk groups have been recognized: (a) low risk disease includes patients with a normal karyotype or isolated del(13q); (b) high risk includes patients with del(11q) and del(17p). Molecular analyses have detected deletions of 13q in cells that are cytogenetically normal as well as abnormal. Heterozygous deletions most frequently occur in the early stage of the disease and homozygous deletions occur in the more advanced stages. In a study examining loss of heterozygosity and subchromosomal copy losses of chromosome 13, two types of deletions were defined: type 1 aberrations occurred in 60% of cases and were associated with loss of Rb1 and breaks close to the miR16/15a locus; and type 2 aberrations that included Rb1 occurred in 40% of cases. The clinical correlation with del(13q) cells include a higher lymphocyte count, a tendency to exhibit a diffuse pattern of bone marrow infiltration, and splenomegaly. They co-exist in only 2% to 5% of patients, suggesting that each change may have a distinct pathogenetic route. Gain of 3q and 8q and loss of 17p are independent unfavorable prognostic biomarkers.

Purchase 250mg meldonium mastercard
Hemoglobin electrophoresis should be performed but should not be relied on as the major diagnostic criterion for ruling in or ruling out a hemoglobinopathy medications 4 less canada purchase meldonium 250 mg on-line. Many amino acid substitutions that have a profound effect on solubility do not change the overall charge on the hemoglobin molecule. This mutation is electrically neutral; it does not alter electrophoretic mobility. A normal electrophoretogram, however, should never be regarded as strong evidence against the presence of a mutant hemoglobin, especially if the clinical picture or family history otherwise supports the diagnosis. Mass spectrometry analysis of hemoglobin and direct globin gene sequencing are supplanting electrophoresis as diagnostic strategies. Additional sophisticated analyses of hemoglobin can be obtained from reference laboratories if detailed characterization seems warranted. For example, abnormal hemoglobin or globin bands migrating to novel positions on an isoelectric focusing gel can result from hemoglobin or globin moieties lacking heme in groups. When heme is added to the sample and the proteins are reanalyzed, these bands disappear. Detection of unstable hemoglobins is occasionally compromised by the selective precipitation of the unstable variant into Heinz bodies. Because most patients are heterozygotes, this phenomenon greatly reduces the apparent percentage of the variant in soluble form. Thus even a variant possessing altered electrophoretic mobility may be very difficult to detect. Indeed, some unstable hemoglobins, such as Hb Geneva or Hb Terre Haute, are so unstable that no mutant gene product can be detected in the steady state. They are detectable only by isotope labeling studies or direct analysis of the globin genes. The amino acid sequence predicted from genetic sequencing may rarely be inaccurate because of posttranslational conversion into an unstable hemoglobin. Through posttranslational modification, the methionine is altered to aspartate, a hydrophilic residue that disrupts the heme pocket. The differential diagnosis of unstable hemoglobin variants is usually straightforward if this general category of hemolytic disorders is suspected. This diagnosis should be considered, as should other causes of chronic or intermittent hemolytic anemia, including red blood cell membrane disorders. Spherocytes are relatively rare in patients with unstable hemoglobin disorders; this is sometimes a useful discriminant. The rate of travel through the various pathways probably differs for the different hemoglobin variants and for a variety of stresses to which the protein is subjected. Many patients can be managed adequately by observation and education to avoid agents that provoke hemolysis. The peripheral smear (A) shows "bite" cells with pitted-out semicircular areas of the red blood cell membrane as a result of removal of Heinz bodies by macrophages in the spleen. The Heinz body preparation (B) shows increased Heinz bodies in the same specimen, when compared to a control (C). Patients who have significant morbidity because of chronic anemia or repeated episodes of severe hemolysis should be considered candidates for splenectomy, especially if hypersplenism has developed. Children with severe hemolysis may require transfusion support until they are old enough (at least 3 or 4 years of age) to undergo splenectomy without unacceptable immunologic compromise. However, splenectomy should be used only as a last resort because of the long-term risks of overwhelming sepsis and thrombosis. Fever should therefore prompt close monitoring of patients for evidence of hemolysis or infection. Postsplenectomy patients with a hemolytic diathesis are also afflicted by a hypercoagulable state, probably due to the deranged membrane architecture resulting from oxidative damage. They thus require monitoring for thrombotic events and may need intermittent or long-term anticoagulant therapy. During the transition from the fully deoxygenated to the fully oxygenated state, the initial oxygenation steps occur with difficulty. In fact, the act of binding the first oxygen molecule increases the affinity of the molecule for subsequent oxygen-binding events, thus creating the sigmoid shape of the curve. The necessary intramolecular reorganization occurs only when the precise arrangement of hydrogen bonds, hydrophobic interactions, and salt bridges is broken and formed in the proper sequence. Mutant hemoglobins exhibiting altered oxygen affinity arise from amino acid substitutions at the interface between - and -chains or in regions affecting the hydrogen bonds, hydrophobic interactions, or salt bridges that influence the interaction of heme with oxygen. These variant hemoglobins cannot acquire any additional oxygen in the lung despite their higher affinity. At normal hematocrit levels, a mild tissue hypoxia results, triggering increased production of erythropoietin and red blood cells, thus resulting in polycythemia. These and numerous other examples that have been analyzed at the molecular level have greatly aided our understanding of the molecular basis for reversible oxygen binding. On the abscissa, the partial pressure of oxygen (Po2) is indicated in millimeters of mercury. On the left ordinate, the saturation of hemoglobin with oxygen is indicated as a percentage; on the right ordinate, the oxygen content of the hemoglobin is expressed as volume percent. Note that the high-affinity hemoglobin delivers less than one-half the oxygen that HbA gives to the tissues, resulting in tissue anoxia, increased erythropoietin secretion, and erythrocytosis. Conversely, the low-affinity hemoglobin is even more efficient than HbA in supplying tissues with oxygen, resulting in diminished erythropoietin production and anemia. The hemoglobin preparation is exposed to increasing oxygen pressures, and the relative percentages of oxyhemoglobin and deoxyhemoglobin are determined. Carbon monoxide stabilizes hemoglobin in the R "oxy" state without the need for oxygen binding. The oxygen affinity curve is therefore extremely left-shifted and is hyperbolic, rather than sigmoidal, in shape. The clinical consequences of mild chronic carbon monoxide poisoning are the same as those seen with high-affinity hemoglobin variants. The most common cause of carbon monoxide toxicity is cigarette smoking, although chronic carbon monoxide exposure can elevate the hematocrit level in people such as caisson workers or tunnel toll collectors. Severe acute carbon monoxide poisoning can cause rapid death as a result of tissue hypoxia. Management Most patients with high-affinity hemoglobins have mild erythrocytosis; they do not require intervention. The blood viscosity is then sufficiently elevated to require therapeutic phlebotomy. When a patient breathes room air, the half-life of carboxyhemoglobin is 4 to 6 hours, but the half-life is 40 to 80 minutes with the use of normobaric oxygen and 15 to 30 minutes with the use of hyperbaric oxygen. Carbon monoxide detectors, designed to detect occult carbon monoxide poisoning, are now required in many municipalities and are predicted to prevent numerous fatalities from occult carbon monoxide poisoning. In cases of Hb Kansas, the threonine position, 102, cannot form a hydrogen bond with aspartic acid at position 94. Most low-affinity variants possess enough oxygen affinity to become fully saturated in the normal lung. First, because tissue oxygen delivery is so "overly" efficient, normal oxygen requirements can be met by lower-than-normal hematocrit levels. This situation produces a state of "pseudoanemia," in which the low hematocrit level is deceiving because both oxygen delivery and the patients are completely normal. Second, the amount of desaturated hemoglobin circulating in capillaries and veins can be greater than 5 g/dL. This usually ominous finding is entirely misleading in these individuals, because it reflects no morbidity. Abnormal hemoglobins producing methemoglobinemia (M hemoglobins) arise from mutations that stabilize the heme iron in the ferric state. Classically a histidine in the vicinity of the heme pocket is replaced by a tyrosine. The oxidized heme iron is relatively resistant to reduction by the methemoglobin reductase system. Methemoglobin has a brownish to blue color that does not revert to red on exposure to oxygen. In contrast to truly cyanotic people, however, arterial partial pressure of oxygen (PaO2) values are usually normal.
Generic meldonium 500 mg fast delivery
Over the past 4 decades medicine rheumatoid arthritis generic 500mg meldonium with amex, clinical experience with deferoxamine, a hexadentate bacterial siderophore purified from Streptomyces pilosus, has established the efficacy and safety of this agent in preventing organ dysfunction and prolonging survival in patients with transfusional iron overload. To be effective, the drug must be administered by prolonged subcutaneous or intravenous infusion with a small portable syringe pump, ideally each day, making compliance a demanding task. In patients with modest iron loads and no evidence of iron toxicity, slow subcutaneous infusion of deferoxamine for 9 to 12 hours daily usually provides adequate therapy. In severely iron-loaded patients and in patients with evidence of iron toxicity, particularly those with cardiac complications, chronic slow intravenous infusions given through an indwelling central venous catheter may permit more rapid reduction of the body iron burden. Deferoxamine is a generally safe and nontoxic drug for iron-loaded patients, but systemic complications have been reported, including allergic anaphylactoid reactions, infectious complications, visual abnormalities, auditory dysfunction, and growth retardation. Adequate deferoxamine therapy should produce a progressive decrease in the body storage iron of almost any patient with iron overload. If no decline is observed, blood and deferoxamine use, compliance, ascorbate status, and other features of the therapeutic regimen should be thoroughly reassessed. Deferasirox, a synthetic, orally active tridentate iron chelator, was approved for use by the U. Food and Drug Administration in 2005 for treatment of transfusional iron overload in adults and in children older than 2 years of age. Brissot P, Ropert M, Le Lan C, et al: Non-transferrin bound iron: a key role in iron overload and iron toxicity. Bowes O, Baxter K, Elsey T, et al: Hereditary hyperferritinaemia cataract syndrome. Additional clinical studies examining the long-term safety and efficacy of deferasirox are in progress, but the initial experience with this drug suggests that this orally active, ironchelating agent is a well-tolerated, once-daily treatment for control of transfusional iron overload. Prognosis the prognosis for patients with iron overload is influenced by many factors, including the magnitude, rate, and route of iron loading; distribution of iron deposition between reticuloendothelial macrophage and parenchymal sites; amount and duration of exposure to circulating nontransferrin-bound iron; ascorbate status; and coexisting disorders, especially alcoholism. Skin pigmentation diminishes; hepatic function may improve while fibrosis is arrested or sometimes regresses; and cardiac abnormalities, including even cardiac failure, may resolve. Diabetes and other endocrine abnormalities usually are ameliorated only slightly, if at all, although reversal of hypogonadism has occurred. Arthropathy usually does not subside and may even continue to progress despite phlebotomy. In patients with iron overload who cannot be treated by phlebotomy, chelation therapy is effective in reducing the body iron burden and improving the prognosis. In all forms of iron overload, the most effective means of preventing complications is prevention of iron accumulation, either by early identification and phlebotomy treatment of hereditary hemochromatosis or by early institution of chelation therapy in patients with iron-loading or transfusion-dependent anemia. Anemia is also extremely common in cancer; 39% of cancer patients were found to be anemic. Chemotherapy with or without radiation therapy increases the incidence of anemia to 63% and 42% respectively. Unlike true dietary iron deficiency, iron is present in the macrophages and total body iron may be normal or elevated. Research over the last decade suggests that synthesis of the iron regulatory antimicrobial peptide hepcidin is induced by inflammatory cytokines during acute and chronic disorders, leading to a state of functional iron deficiency. However, the decrease in life span of red cells is insufficient to explain the degree of anemia. Macrophages play a central role in iron homeostasis and participate in the reutilization of iron from senescent red cells, which supplies more than 95% of the daily iron requirements for normal physiologic processes including erythropoiesis. During inflammation, there is iron retention in the macrophage-phagocytic system and decreased delivery of iron for erythropoiesis. This is consistent with the concept that under inflammatory conditions iron release, but not utilization, is primarily impaired. Activation of immune cells during infections, autoimmune processes, and cancer also leads to the production of numerous cytokines that induce hepcidin production in the liver. Hepcidin binds to the iron export protein ferroportin and interferes with cellular iron export through internalization and degradation of ferroportin. The same mechanism also inhibits iron export from macrophages, aggravating the low serum iron levels3 and enhancing iron retention in the macrophage. Causality is also suggested by the multiple trials of cytokine antagonists in inflammatory diseases that have shown a decrease in anemia. All these studies highlight the importance of cytokines in altering iron metabolism in inflammatory situations. Molecular evidence implicates both a direct and indirect cytokine effect on iron metabolism. Since the increase in ferritin occurs with concomitant downregulation of transferrin receptor expression, the mode of cellular iron sequestration is not entirely clear. Fleming and Sly first suggested a link between hepcidin and abnormalities in iron metabolism with inflammation. Further, it is not uncommon that primary hematologic defects are accompanied by inflammation, or vice versa, so other factors that also result in anemia, such as nutritional deficiency, blood loss, hemolysis, renal failure, and primary bone marrow disorders need to be considered. Iron deficiency can also be secondary to decreased iron absorption, as in autoimmune gastritis and celiac disease. Finally, the contribution of frequent phlebotomy in the hospitalized patients with anemia should not be underestimated (see box on Diagnosis of Anemia of Chronic Diseases). Contributing causes of anemia, including hemolysis, nutritional deficiency, or sequestration should be evaluated. While some investigators argue that a ferritin level greater than 50 ng/ mL excludes any component of iron deficiency even in inflammatory states, a meta-analysis of iron studies in clinical reports from 1842 subjects with serum ferritin levels above 45 ng/mL showed a prevalence of iron deficiency of 6. Other causes of anemia, such as hemolysis, nutritional deficiency, or sequestration, should be ruled out, and a component of iron deficiency should be strongly considered in a patient with systemic inflammation and a low or "normal" serum ferritin concentration. Bone marrow examination is usually not essential for the diagnosis but may be necessary to rule out other diagnoses, including malignancy. Other researchers have shown that, in acute inflammation, serum ferritin levels greater than 3500 ng/mL can coexist with absent bone marrow iron stores tested by aspirate. Although normograms corrected for the degree of inflammation present have been published, most investigators maintain that serum iron studies cannot predictably rule out iron deficiency. However, a bone marrow examination may be necessary to rule out other diagnoses, including iron deficiency, malignancy. However, if the anemia is symptomatic or severe, treatment of the anemia itself may be indicated. However, treatments need to be individualized because the risks of erythropoiesis-stimulating agents or iron therapy in noniron deficient subjects are theoretically real and practically unknowable given the variety of underlying conditions that are incorporated under the rubric of chronic disease. Observations from profoundly anemic subjects without inflammation but religiously opposed to transfusions have suggested a physiologic cutoff for hemoglobin of 5 g/dL, below which increased mortality is seen. In addition, acutely ill patients have not been shown to benefit, in randomized, controlled studies, from transfusion "triggers" above 7 g/dL. Nonetheless, symptomatic improvement is seen in subjects with a range of chronic diseases who are treated for anemia of a more modest degree. Similarly, in cancer subjects referred for radiation therapy, only 16% had hemoglobin levels of less than 10 g/dL. Further, elevations in serum iron have been associated with an increase in cancer risk. Chapter37 AnemiaofChronicDiseases 495 Treatments of anemia in symptomatic patients will need to be individualized, as there are risks with erythropoiesis-stimulating agents as well as iron therapy. However, randomized controlled studies indicate that acutely ill patients do not benefit from transfusion triggers >7 g/dL. More recently, consensus has arisen that, for patients with cancer, erythropoiesis-stimulating agents should be used only in those receiving chemotherapy for palliative, rather than curative, intent. If a hemoglobin response is seen, the dose can be decreased and the interval prolonged to titrate the hemoglobin to an asymptomatic level. Anemia, iron deficiency, and hepcidin may have independent antimicrobial and antitumor roles, and this needs further study. Nairz M, Haschka D, Demetz E, et al: Iron at the interface of immunity and infection. Brugnara C, Schiller B, Moran J: Reticulocyte hemoglobin equivalent (Ret He) and assessment of iron-deficient states.
Generic meldonium 250mg mastercard
There has been little comparative research into methodology for external beam radiation that provides guidance to clinicians symptoms 8-10 dpo cheap 250mg meldonium with mastercard. We recently reviewed our outcomes with a single fraction of external beam radiation, which was hypothesized to optimize convenience and minimize expense to patients. Two hundred and seventy individual lesions in 58 patients were primarily treated with more than 700 cGy (97%). Predictors of poor response included lower extremity lesion, tumors, and lesions exhibiting large-cell transformation. Cost estimates have suggested multifractionated radiation was 200% more expensive than the single fraction. Linear accelerator-generated electron beams are scattered by a penetrable plate placed at the collimator site. Because of this low energy level, the beam only penetrates the surface several millimeters to 1 cm into the dermis. The total skin surface can be treated without significant internal organ toxicity. An excellent review compared results of external beam therapy at Stanford University with those achieved in Hamilton, Ontario (Canada). The results cited in this paper reflect the extensive expertise Chapter85 T-CellLymphomas 1371 of both centers in the delivery of this therapy and may not be applicable to centers where this approach is less frequently used. Ten-year relapse-free survival rates at the two centers ranged from 33% to 52% for this good-prognosis group. Higher risk patients may be "induced" with external beam radiation and then placed on topical chemotherapy or systemic treatments such as extracorporeal photopheresis for "maintenance. Patients with tumor lesions, generalized erythroderma, peripheral blood or nodal involvement, and even visceral spread can be successfully palliated with electron beam radiation therapy as well. Side effects, however, can be occasionally extreme, including scaling, dryness of skin, erythema, edematous extremities, telangiectasia formation, skin ulceration, and hair or sweat gland loss (usually transient but occasionally permanent). Careful radiation dosimetric techniques are required to ensure adequate skin treatment without excessive organ toxicity. Patients have long been known to benefit from the application of topical steroids. Novel approaches by dermatologists to apply chemotherapy topically to avoid systemic therapy complications have also been devised. Although several methods may be used for administration, self-administration at home to the entire skin surface is preferred. The concentration may need to be varied depending on patient tolerance and sensitivity. Several large studies have been completed demonstrating the benefit of mechlorethamine, especially in early-stage disease. One by Vonderheid et al reported their experience with topical mechlorethamine and found that it compared favorably with results achieved with electron beam treatment. The corresponding median duration of remissions was in excess of 15, 5, and 12 months, respectively. It is difficult to draw definite conclusions from these data, because many patients included in the analysis also received intravenous mechlorethamine or methotrexate, and some may have received radiation therapy. Several patients have relapsed as long as 8 years after the completion of therapy, suggesting that follow-up times must be long to compare various topical therapies for early-stage disease. Hoppe et al confirmed these findings using an ointment-based (Aquaphor or polyethylene glycol) topical mechlorethamine, which may be associated with a lower incidence of cutaneous sensitivity. It is also quick drying, greaseless, and developed under good manufacturing practices (not compounded in pharmacy). For early-stage disease, topical mechlorethamine offers an efficient, convenient (outpatient treatment), and relatively inexpensive treatment option. Side effects consist of delayed hypersensitivity in approximately 35% of patients, although ointment-based solutions appeared to offer a reduced risk for allergic contact dermatitis. Once hypersensitivity develops, patients can be desensitized by injecting minute daily doses of mechlorethamine over a period of several weeks. This should be done only in a medical setting with appropriate anaphylaxis precautions observed. Other investigators had difficulty replicating the results using this risky procedure, and topical desensitization has become much more common. Constantine et al propose that therapy should be transiently discontinued until clearance of the allergic dermatitis is achieved (using topical steroids if necessary). If this dilution is tolerated, the dosage is doubled weekly until the dose achieved is often identical to the initial concentration that induced the hypersensitivity. Some clinicians, however, believe that a mild hypersensitivity reaction may have beneficial antitumor effects. Some hypersensitivity may be beneficial; the generalized erythroderma and pruritus are usually poorly tolerated when severe, however, and some alteration in therapy is required. An increased risk for secondary skin cancers in patients receiving long-term mechlorethamine has been observed. Some physicians have expressed concern regarding the safety of family members or healthcare workers secondarily exposed to the topical solutions. Home treatment with topical mechlorethamine has been shown to result in aerosolized drug levels, which may result in mucous membrane or ocular irritation. However, we are unaware of any documented adverse outcomes and believe this to be a theoretical concern rather than a practical one. However, topical carmustine is the only one of these agents that has demonstrated clinical use. A stock solution is created with 300 mg carmustine in 150 mL of 95% ethanol (sufficient for 30 days of treatment). This solution can then be applied to the general body surface, with the exception of the head, genitals, palms, soles, and intertriginous zones, unless involved by disease. For more extensive patch-stage disease, the freedom from treatment failure rate at 3 years was 62%. Side effects of contact dermatitis are less frequent with this agent, but systemic side effects, mainly leukopenia, are more common. This drug may be helpful for the treatment of patients who do not tolerate topical mechlorethamine, because there is no crosssensitivity. Hamminga et al performed one of the few prospective trials comparing total-skin electron beam radiation to topical mechlorethamine. Patients were not randomized to their treatment; rather, the physician based the decision on patient health, availability of the linear accelerator, and the distance the patient lived from the clinic. In more advanced skin disease, a trend toward superior initial response was seen with electron beam therapy, but there was a high relapse rate in those patients, necessitating subsequent therapy. Systemic Chemotherapy Currently systemic chemotherapy is reserved for those patients with relapsed or refractory disease after topical interventions or for those patients with advanced nodal or visceral disease at presentation. Many of the patients treated with chemotherapy have also previously been treated with cytokine-based or other nontraditional chemotherapeutic agents before systemic chemotherapy is considered. Vorinostat 400 mg daily orally was tested in an open-label trial of 74 patients who had progressed on at least two prior systemic therapies. Grade 3 events were less common but included fatigue (5%), deep venous thromboses/pulmonary emboli (5%), and thrombocytopenia (4%). Common adverse events included nausea/vomiting in over 56% of patients, asthenia in 44%, and diarrhea in 14%, but again grade 3 toxicities were much less frequent, but also included fatigue in 6%. However, in this large trial, events such as cardiac failure, atrioventricular block, and ventricular tachycardia were rare, occurring typically in less than 1% of patients. The second confirmatory trial was led by investigators at the National Cancer Institute. In general then, toxicity to romidepsin and vorinostat has included alterations in the cardiac conduction that could potentially predispose to arrhythmias, and treatment of patients has required ongoing telemetry monitoring in some trials, but no evidence for acute or chronic impairment in cardiac function has been noted. Vorinostat therapy led to drug-related grade 1 electrocardiographic changes in five patients and grade 2 in one patient. McDonald and Bertino reported particularly good results with the antimetabolite methotrexate administered intravenously followed by oral citrovorum factor.
