

Purchase 0.1mg florinef fast delivery
It can be given by deep intramuscular injection or by intravenous infusion gastritis quizlet buy florinef overnight delivery, although the intravenous route is used most commonly. Intravenous administration eliminates the local pain and tissue staining that often occur with the intramuscular route and allows delivery of the entire dose of iron necessary to correct the iron deficiency at one time. Adverse effects of intravenous iron dextran therapy include headache, light-headedness, fever, arthralgias, nausea and vomiting, back pain, flushing, urticaria, bronchospasm, and, rarely, anaphylaxis and death. Owing to the risk of a hypersensitivity reaction, a small test dose of iron dextran should always be given before full intramuscular or intravenous doses are given. Patients with a strong history of allergy and patients who have previously received parenteral iron dextran are more likely to have hypersensitivity reactions after treatment with parenteral iron dextran. The iron dextran formulations used clinically are distinguishable as high-molecular-weight and low-molecular-weight forms. Clinical data-primarily from observational studies-indicate that the risk of anaphylaxis is largely associated with high-molecular-weight formulations. Sodium ferric gluconate complex and iron-sucrose complex are alternative parenteral iron preparations. Ferric carboxymaltose is a colloidal iron preparation embedded within a carbohydrate polymer. Ferumoxytol is a superparamagnetic iron oxide nanoparticle coated with carbohydrate. The carbohydrate shell is removed in the reticuloendothelial system, allowing the iron to be stored as ferritin, or released to transferrin. For patients treated chronically with parenteral iron, it is important to monitor iron storage levels to avoid the serious toxicity associated with iron overload. Unlike oral iron therapy, which is subject to the regulatory mechanism provided by the intestinal uptake system, parenteral administration-which bypasses this regulatory system-can deliver more iron than can be safely stored. Although deficiency of vitamin B12 due to an inadequate supply in the diet is unusual, deficiency of B12 in adults-especially older adults-due to inadequate absorption of dietary vitamin B12 is a relatively common and easily treated disorder. Acute Iron Toxicity Acute iron toxicity is seen almost exclusively in young children who accidentally ingest iron tablets. As few as 10 tablets of any of the commonly available oral iron preparations can be lethal in young children. Adult patients taking oral iron preparations should be instructed to store tablets in child-proof containers out of the reach of children. Children who are poisoned with oral iron experience necrotizing gastroenteritis with vomiting, abdominal pain, and bloody diarrhea followed by shock, lethargy, and dyspnea. Subsequently, improvement is often noted, but this may be followed by severe metabolic acidosis, coma, and death. Whole bowel irrigation (see Chapter 58) should be performed to flush out unabsorbed pills. Deferoxamine, a potent iron-chelating compound, can be given intravenously to bind iron that has already been absorbed and to promote its excretion in urine and feces. Activated charcoal, a highly effective adsorbent for most toxins, does not bind iron and thus is ineffective. Appropriate supportive therapy for gastrointestinal bleeding, metabolic acidosis, and shock must also be provided. Chronic Iron Toxicity Chronic iron toxicity (iron overload), also known as hemochromatosis, results when excess iron is deposited in the heart, liver, pancreas, and other organs. It most commonly occurs in patients with inherited hemochromatosis, a disorder characterized by excessive iron absorption, and in patients who receive many red cell transfusions over a long period of time (eg, individuals with -thalassemia). Chronic iron overload in the absence of anemia is most efficiently treated by intermittent phlebotomy. One unit of blood can be removed every week or so until all of the excess iron is removed. Iron chelation therapy using parenteral deferoxamine or the oral iron chelators deferasirox or deferiprone (see Chapter 57) is less efficient as well as more complicated, expensive, and hazardous, but it may be the only option for iron overload that cannot be managed by phlebotomy, as is the case for many individuals with inherited and acquired causes of refractory anemia such as thalassemia major, Chemistry Vitamin B12 consists of a porphyrin-like ring with a central cobalt atom attached to a nucleotide. Various organic groups may be covalently bound to the cobalt atom, forming different cobalamins. Deoxyadenosylcobalamin and methylcobalamin are the active forms of the vitamin in humans. Cyanocobalamin and hydroxocobalamin (both available for therapeutic use) and other cobalamins found in food sources are converted to the active forms. The ultimate source of vitamin B12 is from microbial synthesis; the vitamin is not synthesized by animals or plants. The chief dietary source of vitamin B12 is microbially derived vitamin B12 in meat (especially liver), eggs, and dairy products. Vitamin B12 is sometimes called extrinsic factor to differentiate it from intrinsic factor, a protein secreted by the stomach that is required for gastrointestinal uptake of dietary vitamin B12. Because the normal daily requirements of vitamin B12 are only about 2 mcg, it would take about 5 years for all of the stored vitamin B12 to be exhausted and for megaloblastic anemia to develop if B12 absorption were stopped. Vitamin B12 is absorbed after it complexes with intrinsic factor, a glycoprotein secreted by the parietal cells of the gastric mucosa. Vitamin B12 deficiency in humans most often results from malabsorption of vitamin B12 due either to lack of intrinsic factor or to loss or malfunction of the absorptive mechanism in the distal ileum. Nutritional deficiency is rare but may be seen in strict vegetarians after many years without meat, eggs, or dairy products. Without vitamin B12, conversion of the major dietary and storage folate-N 5-methyltetrahydrofolate-to tetrahydrofolate, the precursor of folate cofactors, cannot occur. As a result, vitamin B12 deficiency leads to deficiency of folate cofactors necessary for several biochemical reactions involving the transfer of one-carbon groups. The accumulation of folate as N 5-methyltetrahydrofolate and the associated depletion of tetrahydrofolate cofactors in vitamin B12 deficiency have been referred to as the "methylfolate trap. Methyl transfer N 5-Methyltetrahydrofolate Tetrahydrofolate Cobalamin Methylcobalamin Methionine B. Section 3 shows the pathway by which folic acid enters the tetrahydrofolate cofactor pool. There is evidence from observational studies that elevated serum homocysteine increases the risk of atherosclerotic cardiovascular disease. However, randomized clinical trials have not shown a definitive reduction in cardiovascular events (myocardial infarction, stroke) in patients receiving vitamin supplementation that lowers serum homocysteine. In vitamin B12 deficiency, this conversion cannot take place and the substrate, methylmalonyl-CoA, as well as methylmalonic acid accumulate. In the past, it was thought that abnormal accumulation of methylmalonyl-CoA causes the neurologic manifestations of vitamin B12 deficiency. However, newer evidence implicates the disruption of the methionine synthesis pathway as the cause of neurologic problems. Whatever the biochemical explanation for neurologic damage, the important point is that administration of folic acid in the setting of vitamin B12 deficiency will not prevent neurologic manifestations even though it will largely correct the anemia caused by the vitamin B12 deficiency. The neurologic syndrome associated with vitamin B12 deficiency usually begins with paresthesias in peripheral nerves and weakness and progresses to spasticity, ataxia, and other central nervous system dysfunctions. Correction of vitamin B12 deficiency arrests the progression of neurologic disease, but it may not fully reverse neurologic symptoms that have been present for several months.
Discount florinef 0.1mg mastercard
In contrast to methadone gastritis peptic ulcers symptoms buy florinef in united states online, high-dose administration of buprenorphine results in a -opioid antagonist action, limiting its properties of analgesia and respiratory depression. Buprenorphine is also available combined with naloxone, a pure -opioid antagonist (as Suboxone), to help prevent its diversion for illicit intravenous misuse. A slow-release transdermal patch preparation that releases drug over a 1-week period is also available (Butrans). Psychotomimetic effects, with hallucinations, nightmares, and anxiety, have been reported after use of drugs with mixed agonist-antagonist actions. Pentazocine (a benzomorphan) and nalbuphine are other examples of opioid analgesics with mixed agonist-antagonist properties. Nalbuphine is a strong -receptor agonist and a partial -receptor antagonist; it is given parenterally. At higher doses there seems to be a definite ceiling-not noted with morphine-to the respiratory depressant effect. Unfortunately, when respiratory depression does occur, it may be relatively resistant to naloxone reversal due to its greater affinity for the receptor than naloxone. Nalbuphine is equipotent to morphine for analgesia and, at lower doses, can be effective for pruritus for opioid and nonopioid etiologies. Morphinans Butorphanol produces analgesia equivalent to nalbuphine but appears to produce more sedation at equianalgesic doses. Benzomorphans Pentazocine is a agonist with weak -antagonist or partial agonist properties. However, because of its irritant properties, the injection of pentazocine subcutaneously is not recommended. Because its analgesic effect is only partially antagonized by naloxone, it is thought to depend less on its low-affinity binding to the receptor for therapeutic activity. Toxicity includes association with seizures; the drug is relatively contraindicated in patients with a history of epilepsy and for use with other drugs that lower the seizure threshold. Another serious risk is the development of serotonin syndrome, especially if selective serotonin reuptake inhibitor antidepressants are being administered (see Chapter 16). Other adverse effects include nausea and dizziness, but these symptoms typically abate after several days of therapy. No clinically significant effects on respiration or the cardiovascular system have thus far been reported when used as monotherapy. Given the fact that the analgesic action of tramadol is largely independent of -receptor action, tramadol may serve as an adjunct with pure opioid agonists in the treatment of chronic neuropathic pain. Tapentadol is an analgesic with modest -opioid receptor affinity and significant norepinephrine reuptake-inhibiting action. In animal models, its analgesic effects were only moderately reduced by naloxone but strongly reduced by an 2-adrenoceptor antagonist. Tapentadol was approved in 2008 and has been shown to be as effective as oxycodone in the treatment of moderate to severe pain but with a reduced profile of gastrointestinal complaints such as nausea. Tapentadol carries risk for seizures in patients with seizure disorders and for the development of serotonin syndrome. It is unknown how tapentadol compares in clinical utility to tramadol or other analgesics whose mechanism of action is not based primarily on opioid receptor pharmacology. Moreover, because of variations in the metabolism of codeine, its use for any purpose in young children is being reconsidered. Dextromethorphan is the dextrorotatory stereoisomer of a methylated derivative of levorphanol. It is purported to be free of addictive properties and produces less constipation than codeine. Dextromethorphan has also been found to enhance the analgesic action of morphine and presumably other -receptor agonists. However, misuse of its purified (powdered) form has been reported to lead to serious adverse events including death. Codeine, as noted, has a useful antitussive action at doses lower than those required for analgesia. Levopropoxyphene is the stereoisomer of the weak opioid agonist dextropropoxyphene. It is devoid of opioid effects, although sedation has been described as a side effect. They have lower affinity for the other receptors but can also reverse agonists at and sites. This effect is often achieved at doses below those necessary to produce analgesia. The receptors involved in the antitussive effect appear to differ from those associated with the other actions of opioids. For example, the antitussive effect is also produced by stereoisomers of opioid molecules that are devoid of analgesic effects and addiction liability (see below). The physiologic mechanism of cough is complex, and little is known about the specific mechanism of action of the opioid antitussive drugs. Metabolic disposition is chiefly by glucuronide conjugation like that of the agonist opioids with free hydroxyl groups. Naltrexone is well absorbed after oral administration but may undergo rapid firstpass metabolism. It has a half-life of 10 hours, and a single oral dose of 100 mg blocks the effects of injected heroin for up to 48 hours. Nalmefene, the newest of these agents, is a derivative of naltrexone but is available only for intravenous administration. In individuals who are acutely depressed by an overdose of an opioid, the antagonist effectively normalizes respiration, level of consciousness, pupil size, bowel activity, and awareness of pain. In dependent subjects who appear normal while taking opioids, naloxone or naltrexone almost instantaneously precipitates an abstinence syndrome. There is no tolerance to the antagonistic action of these agents, nor does withdrawal after chronic administration precipitate an abstinence syndrome. Clinical Use Naloxone is a pure antagonist and is preferred over older weak agonist-antagonist agents that had been used primarily as antagonists, eg, nalorphine and levallorphan. The major application of naloxone is in the treatment of acute opioid overdose (see also Chapter 58). Careful titration of the naloxone dosage can often eliminate the itching, nausea, and vomiting while sparing the analgesia. Methylnaltrexone has a quaternary amine preventing it from crossing the blood-brain barrier. Alvimopan has a high affinity for peripheral receptors and does not impair the central effects of -opioid agonists. Because of its long duration of action, naltrexone has been proposed as a maintenance drug for addicts in treatment programs. A single dose given on alternate days blocks virtually all of the effects of a dose of heroin. It might be predicted that this approach to rehabilitation would not be popular with a large percentage of drug users unless they are motivated to become drugfree. Naltrexone also facilitates abstinence from nicotine (cigarette smoking) with reduced weight gain. In fact, a combination of naltrexone plus bupropion (Chapter 16) may also offer an effective and synergistic strategy for weight loss.
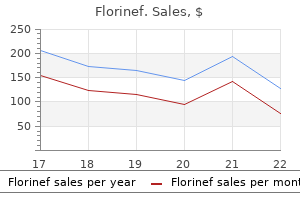
| Comparative prices of Florinef | ||
| # | Retailer | Average price |
| 1 | Foot Locker | 906 |
| 2 | Gap | 243 |
| 3 | Ahold USA / Royal Ahold | 965 |
| 4 | Big Lots | 753 |
| 5 | Bed Bath & Beyond | 603 |
| 6 | Menard | 584 |
Florinef 0.1mg with mastercard
Sotalol is approved for the treatment of life-threatening ventricular arrhythmias and the maintenance of sinus rhythm in patients with atrial fibrillation gastritis symptoms palpitations purchase cheapest florinef and florinef. It is also approved for treatment of supraventricular and ventricular arrhythmias in the pediatric age group. Intravenous ibutilide is used for the acute conversion of atrial flutter and atrial fibrillation to normal sinus rhythm. The drug is more effective in atrial flutter than atrial fibrillation, with a mean time to termination of 20 minutes. The dihydropyridines (eg, nifedipine) do not share antiarrhythmic efficacy and may precipitate arrhythmias. Dofetilide produces no relevant blockade of the other potassium channels or the sodium channel. Because of the slow rate of recovery from blockade, the extent of blockade shows little dependence on stimulation frequency. Verapamil increases peak plasma dofetilide concentration by increasing intestinal blood flow. Eighty percent of an oral dose is eliminated unchanged by the kidneys; the remainder is eliminated in the urine as inactive metabolites. Inhibitors of the renal cation secretion mechanism, eg, cimetidine, prolong the half-life of dofetilide. Dofetilide is approved for the maintenance of normal sinus rhythm in patients with atrial fibrillation. It is also effective in restoring normal sinus rhythm in patients with atrial fibrillation. Verapamil can suppress both early and delayed afterdepolarizations and may abolish slow responses arising in severely depolarized tissue. Extracardiac Effects Verapamil causes peripheral vasodilation, which may be beneficial in hypertension and peripheral vasospastic disorders. Its effects on smooth muscle produce a number of extracardiac effects (see Chapter 12). A common error has been to administer intravenous verapamil to a patient with ventricular tachycardia misdiagnosed as supraventricular tachycardia. Adverse extracardiac effects include constipation, lassitude, nervousness, and peripheral edema. Activation of slow inward sodium current has also been suggested as an additional mechanism of action potential prolongation. It is extensively metabolized by the liver; after oral administration, its bioavailability is only about 20%. Therefore, verapamil must be administered with caution in patients with hepatic dysfunction or impaired hepatic perfusion. Effective oral dosages are higher than intravenous dosage because of first-pass metabolism and range from 120 mg to 640 mg daily, divided into three or four doses. It is also becoming clear that certain nonantiarrhythmic drugs, such as drugs acting on the renin-angiotensin-aldosterone system, fish oil, and statins, can reduce recurrence of tachycardias and fibrillation in patients with coronary heart disease or congestive heart failure. Its cardiac mechanism of action involves activation of an inward rectifier K+ current and inhibition of calcium current. The results of these actions are marked hyperpolarization and suppression of calciumdependent action potentials. It is usually given in a bolus dose of 6 mg followed, if necessary, by a dose of 12 mg. The drug is less effective in the presence of adenosine receptor blockers such as theophylline or caffeine, and its effects are potentiated by adenosine uptake inhibitors such as dipyridamole. Therapeutic Use Supraventricular tachycardia is the major arrhythmia indication for verapamil. Adenosine or verapamil is preferred over older treatments (propranolol, digoxin, edrophonium, vasoconstrictor agents, and cardioversion) for termination. Verapamil can also reduce the ventricular rate in atrial fibrillation and flutter ("rate control"). However, intravenous verapamil in a patient with sustained ventricular tachycardia can cause hemodynamic collapse. An intravenous form of diltiazem is available for the latter indication and causes hypotension or bradyarrhythmias relatively infrequently. The Nonpharmacologic Therapy of Cardiac Arrhythmias It was recognized over 100 years ago that reentry in simple in vitro models (eg, rings of conducting tissues) was permanently interrupted by transecting the reentry circuit. This concept is now applied in cardiac arrhythmias with defined anatomic pathways-eg, atrioventricular reentry using accessory pathways, atrioventricular node reentry, atrial flutter, and some forms of ventricular tachycardia-by treatment with radiofrequency catheter ablation or extreme cold, cryoablation. Mapping of reentrant pathways and ablation can be carried out by means of catheters threaded into the heart from peripheral arteries and veins. Studies have also shown that paroxysmal and persistent atrial fibrillation may arise from one or more of the pulmonary veins. Both forms of atrial fibrillation can be cured by electrically isolating the pulmonary veins by radiofrequency or cryotherapy catheter ablation or during concomitant cardiac surgery. Because catheter ablation therapy can often permanently cure atrial fibrillation, and because it does not involve adverse effects of drugs, it has become a very common treatment for chronic atrial fibrillation. The increasing use of nonpharmacologic antiarrhythmic therapies reflects both advances in the relevant technologies and an increasing appreciation of the dangers of long-term therapy with currently available drugs. It slows pacemaker activity by decreasing diastolic depolarization of sinus node cells. Unlike other heart rate-lowering agents such as blockers, it reduces heart rate without affecting myocardial contractility, ventricular repolarization, or intracardiac conduction. Elevated heart rate is an important determinant of the ischemic threshold in patients with coronary artery disease and a prognostic indicator in patients with congestive heart failure. Antianginal and anti-ischemic effects of ivabradine have been demonstrated in patients with coronary artery disease and chronic stable angina. In controlled clinical trials, ivabradine proved as effective as blockers in the control of angina. In patients with left ventricular dysfunction and heart rates greater than 70 bpm, ivabradine reduced mean heart rate and the composite end points of cardiovascular mortality and hospitalization. Inappropriate sinus tachycardia is an uncommon disorder characterized by multiple symptoms, including palpitations, dizziness, orthostatic intolerance, and elevated heart rates. Conventional treatment includes blockers and nondihydropyridine calcium channel blockers. Recent case reports and one clinical trial have shown that ivabradine provides an effective alternative to slow the heart rate in patients with inappropriate sinus tachycardia. Visual disturbances attributable to the block of the If channels in the retina have been described. This side effect is limited by the low permeability of ivabradine in the blood-brain barrier. Ranolazine had been shown to have antiarrhythmic properties in both atrial and ventricular arrhythmias. It is currently undergoing clinical trials in combination with dronedarone for the suppression of atrial fibrillation. Ranolazine has been shown to suppress ventricular tachycardia in ischemic models and in a major clinical trial of its effects in coronary artery disease. It causes frequency- and voltagedependent block of the early and late components of the sodium current. The early-activating potassium channels Ito and Ikur are also blocked by the drug. These potassium channel currents play a more prominent role in atrial than ventricular repolarization. In a direct comparison trial, vernakalant proved more effective than placebo or amiodarone in terminating atrial fibrillation in a 90-minute period. This relatively rapid action decreases the required observation period for untoward side effects following drug administration. Sinus bradycardia and hypotension are the only noticeable cardiovascular adverse effects. Magnesium therapy appears to be indicated in patients with digitalis-induced arrhythmias if hypomagnesemia is present; it is also indicated in some patients with torsades de pointes even if serum magnesium is normal.
Discount florinef 0.1mg amex
Capsaicin causes the release of the peptide substance P from sensory neurons gastritis upper left abdominal pain florinef 0.1mg on-line, and tetanus toxin blocks the release of transmitters. For most neurotransmitters, there are uptake mechanisms into the synaptic terminal and also into surrounding neuroglia. Cocaine, for example, blocks the uptake of catecholamines at adrenergic synapses and thus potentiates the action of these amines. Anticholinesterases block the degradation of acetylcholine and thereby prolong its action (see Chapter 7). In the postsynaptic region, the transmitter receptor provides the primary site of drug action. Drugs can act either as neurotransmitter agonists, such as the opioids, which mimic the action of enkephalin, or they can block receptor function. In the case of metabotropic receptors, drugs can act at any of the steps downstream of the receptor. The traditional view of the synapse is that it functions like a valve, transmitting information in one direction. However, it is now clear that the synapse can generate signals that feed back onto the presynaptic terminal to modify transmitter release. Endocannabinoids are the best documented example of such retrograde signaling (see below). First, with a few exceptions, different neurotransmitters are released by different groups of neurons. For example, there are at least 14 different serotonin receptors encoded by different genes. E Relay neurons Hierarchical Systems Hierarchical systems include all the pathways directly involved in sensory perception and motor control. These pathways are generally clearly delineated, being composed of large myelinated fibers that can often conduct action potentials at a rate of more than 50 m/s. In sensory systems, the information is processed sequentially by successive integrations at each relay nucleus on its way to the cortex. The projection neurons form the interconnecting pathways that transmit signals over long distances. Their cell bodies are relatively large, and their axons can project long distances but also emit small collaterals that synapse onto local interneurons. These neurons are excitatory, and their synaptic influences, which involve ionotropic receptors, are very shortlived. The excitatory transmitter released from these cells is, in most instances, glutamate. Local circuit neurons are typically smaller than projection neurons, and their axons arborize in the immediate vicinity of the cell body. They synapse primarily on the cell body of the projection neurons but can also synapse on the dendrites of projection neurons as well as with each other. A shows parts of three excitatory relay neurons (blue) and two types of local inhibitory interneuron pathways, recurrent and feed-forward. B shows the pathway responsible for axoaxonic presynaptic inhibition in which the axon of an inhibitory neuron (gray) synapses onto the presynaptic axon terminal of an excitatory fiber (blue) to inhibit its neurotransmitter release. Nonspecific or Diffuse Neuronal Systems Neuronal systems containing many of the other neurotransmitters, including the monoamines and acetylcholine, differ in fundamental ways from the hierarchical systems. For example, noradrenergic cell bodies are found primarily in a compact cell group called the locus coeruleus located in the caudal pontine central gray matter and number only approximately 12,000 neurons on each side of the human brain. Because the axons from these diffusely projecting neurons are fine and unmyelinated, they conduct very slowly, at about 0. In the neocortex, these fibers have a tangential organization and therefore can influence large areas of cortex. In addition, most neurotransmitters utilized by diffuse neuronal systems, including norepinephrine, act predominantly on metabotropic receptors and therefore initiate longlasting synaptic effects. It is not surprising, then, that these systems have been implicated in such global functions as sleeping and waking, attention, appetite, and emotional states. Other diffusely projecting neurotransmitter pathways include the histamine and orexin systems (not shown). Establishing that a chemical substance is a transmitter has been far more difficult for central synapses than for peripheral synapses. Localization: A suspected transmitter must reside in the presynaptic terminal of the pathway of interest. Release: A suspected transmitter must be released from a neuron in response to neuronal activity and in a calcium-dependent manner. Synaptic Mimicry: Application of the candidate substance should produce a response that mimics the action of the transmitter released by nerve stimulation, and application of a selective antagonist should block the response. Glutamate Excitatory synaptic transmission is mediated by glutamate, which is present in very high concentrations in excitatory synaptic vesicles (~100 mM). In glia, glutamate is converted to glutamine by glutamine synthetase, released from the glia, taken up by the nerve terminal, and converted back to glutamate by the enzyme glutaminase. This excitation is caused by the activation of both ionotropic and metabotropic receptors, which have been extensively characterized by molecular cloning. Consumption of contaminated shellfish has been implicated in illness in animals and humans. In addition to glutamate binding, the channel also requires the binding of glycine to a separate site in order for the channel to open. Only when the neuron is strongly depolarized, as occurs with intense activation of the synapse or by activation of neighboring synapses, is the Mg2+ expelled, allowing the channel to open. This enhancement of synaptic strength, which is one major type of synaptic plasticity, can last for many hours or even days and is generally accepted as an important cellular mechanism underlying learning and memory. The metabotropic glutamate receptors are G protein-coupled receptors that act indirectly on ion channels via G proteins. A variety of agonists and antagonists have been developed that interact selectively with the different groups. Group I receptors are typically located postsynaptically and activate phospholipase C, leading to inositol trisphosphate-mediated intracellular Ca2+ release. Activation of these receptors causes the inhibition of Ca2+ channels, resulting in inhibition of transmitter release. These receptors are activated only when the concentration of glutamate rises to high levels during repetitive stimulation of the synapse. Glutamine is imported into the glutamatergic neuron (A) and converted into glutamate by glutaminase. Synaptic transmission is terminated by active transport of the glutamate into a neighboring glial cell (C) by a glutamate transporter. It is converted into glutamine by glutamine synthetase and transported back into the glutamatergic axon terminal. Strychnine, which is a potent spinal cord convulsant and has been used in some rat poisons, selectively blocks glycine receptors. Inhibitory postsynaptic potentials in many areas of the brain have a fast and slow component. The difference in kinetics stems from the differences in coupling of the receptors to ion channels. These receptors are selectively inhibited by picrotoxin and bicuculline, both of which cause generalized convulsions. These receptors are coupled to G proteins that, depending on their cellular location, either inhibit Ca2+ channels or activate K+ channels. This inhibitory postsynaptic potential is long-lasting and slow because the coupling of receptor activation to K+ channel opening is indirect and delayed. A far more widespread muscarinic action in response to acetylcholine is a slow excitation that in some cases is mediated by M1 receptors. These muscarinic effects are much slower than either nicotinic effects on Renshaw cells or the effect of amino acids. Furthermore, this M1 muscarinic excitation is unusual in that acetylcholine produces it by decreasing the membrane permeability to potassium, ie, the opposite of conventional transmitter action. These include neurons in the neostriatum, the medial septal nucleus, and the reticular formation that appear to play an important role in cognitive functions, especially memory. Presenile dementia of the Alzheimer type is reportedly associated with a profound loss of cholinergic neurons.
Order cheapest florinef and florinef
It has less troublesome adverse effects than reserpine gastritis diet �������� buy generic florinef line, which has also been used for this purpose. For poor metabolizers, the maximum recommended dose is 50 mg daily (25 mg/dose); otherwise, a maximum dose of 100 mg daily can be used. Treatment with postsynaptic dopamine receptor blockers such as phenothiazines and butyrophenones may also be helpful. Haloperidol is started in a small dose, eg, 1 mg twice daily, and increased every 4 days depending on the response. If haloperidol is not helpful, treatment with increasing doses of fluphenazine in a similar dose, eg, 1 mg twice daily, sometimes helps. Several recent reports suggest that olanzapine may also be useful; the dose varies with the patient, but 10 mg daily is often sufficient, although doses as high as 30 mg daily are sometimes required. The pharmacokinetics and clinical properties of these drugs are considered in greater detail elsewhere in this book. Selective serotonin reuptake inhibitors may reduce depression, aggression, and agitation. Deutetrabenazine is contraindicated in patients on monoamine oxidase inhibitors, reserpine, or tetrabenazine, and in those who are severely depressed or suicidal. Other important aspects of management include genetic counseling, speech therapy, physical and occupational therapy, dysphagia precautions, and provision of social services. Treatment is directed at the underlying cause when chorea occurs as a complication of general medical disorders such as thyrotoxicosis, polycythemia vera rubra, systemic lupus erythematosus, hypocalcemia, and hepatic cirrhosis. Drug-induced chorea is managed by withdrawal of the offending substance, which may be levodopa, an antimuscarinic drug, amphetamine, lithium, phenytoin, or an oral contraceptive. Neuroleptic drugs may also produce an acute or tardive dyskinesia (discussed below). Ballismus the biochemical basis of ballismus is unknown, but the pharmacologic approach to management is the same as for chorea. Treatment with tetrabenazine, haloperidol, perphenazine, or other dopamine-blocking drugs may be helpful. Athetosis & Dystonia the physiologic basis of these disorders is unknown, and there is no satisfactory medical treatment for them. A subset of patients respond well to levodopa medication (dopa-responsive dystonia), which is therefore worthy of trial. Occasional patients with dystonia may respond to diazepam, amantadine, antimuscarinic drugs (in high dosage), carbamazepine, baclofen, haloperidol, or phenothiazines. A trial of these pharmacologic approaches is worthwhile, though often not successful. Patients with focal dystonias such as blepharospasm or torticollis often benefit from injection of botulinum toxin into the overactive muscles. The role of repetitive transcranial magnetic stimulation and transcranial direct current stimulation to induce plastic changes in the brain is being explored. Pharmacologic therapy may be necessary when tics interfere with social life or otherwise impair activities of daily living. Treatment is with drugs that block dopamine receptors or deplete dopamine stores, such as fluphenazine, pimozide, and tetrabenazine. Pimozide, a dopamine receptor antagonist, may be helpful in patients as a first-line treatment or in those who are either unresponsive to or intolerant of the other agents Other Forms of Chorea Benign hereditary chorea is inherited (usually autosomal dominant; possibly also autosomal recessive) or arises spontaneously. Chorea develops in early childhood and does not progress during adult life; dementia does not occur. It has similar side effects to haloperidol but may cause irregularities of cardiac rhythm. Patients are better able to tolerate this drug if treatment is started with a small dosage (eg, 0. Adverse effects include extrapyramidal movement disorders, sedation, dryness of the mouth, blurred vision, and gastrointestinal disturbances. The most common adverse effect is sedation; other adverse effects include reduced or excessive salivation and diarrhea. Both of these drugs may be particularly helpful for behavioral symptoms, such as impulse control disorders. Atypical antipsychotics, such as risperidone and aripiprazole, may be especially worthwhile in patients with significant behavioral problems. Injection of botulinum toxin A at the site of problematic tics is sometimes helpful when these are focal simple tics. Treatment of any associated attention deficit disorder (eg, with clonidine patch, guanfacine, pemoline, methylphenidate, or dextroamphetamine) or obsessive-compulsive disorder (with selective serotonin reuptake inhibitors or clomipramine) may be required. Chorea may also develop in patients receiving phenytoin, carbamazepine, amphetamines, lithium, and oral contraceptives, and it resolves with discontinuance of the offending medication. Dystonia has resulted from administration of dopaminergic agents, lithium, serotonin reuptake inhibitors, carbamazepine, and metoclopramide; and postural tremor from theophylline, caffeine, lithium, valproic acid, thyroid hormone, tricyclic antidepressants, and isoproterenol. The pharmacologic basis of the acute dyskinesia or dystonia sometimes precipitated by the first few doses of a phenothiazine is not clear. Tardive dyskinesia, a disorder characterized by a variety of abnormal movements, is a common complication of long-term neuroleptic or metoclopramide drug treatment (see Chapter 29). A reduction in dose of the offending medication, a dopamine receptor blocker, commonly worsens the dyskinesia, whereas an increase in dose may suppress it. The drugs most likely to provide immediate symptomatic benefit are those interfering with dopaminergic function, either by depletion (eg, reserpine, tetrabenazine) or receptor blockade (eg, phenothiazines, butyrophenones). Paradoxically, the receptor-blocking drugs are the ones that also cause the dyskinesia. Tardive dystonia is usually segmental or focal; generalized dystonia is less common and occurs in younger patients. Treatment is the same as for tardive dyskinesia, but anticholinergic drugs may also be helpful; focal dystonias may also respond to local injection of botulinum A toxin. Rabbit syndrome, another neuroleptic-induced disorder, is manifested by rhythmic vertical movements about the mouth; it may respond to anticholinergic drugs. Because the tardive syndromes that develop in adults are often irreversible and have no satisfactory treatment, care must be taken to reduce the likelihood of their occurrence. Antipsychotic medication should be prescribed only when necessary and should be withheld periodically to assess the need for continued treatment and to unmask incipient dyskinesia. Thioridazine, a phenothiazine with a piperidine side chain, is an effective antipsychotic agent that seems less likely than most to cause extrapyramidal reactions, perhaps because it has little effect on dopamine receptors in the striatal system. Finally, antimuscarinic drugs should not be prescribed routinely in patients receiving neuroleptics, because the combination may increase the likelihood of dyskinesia. Treatment includes withdrawal of antipsychotic drugs, lithium, and anticholinergics; reduction of body temperature; and rehydration. Dantrolene, dopamine agonists, levodopa, or amantadine may be helpful, but there is a high mortality rate (up to 20%) with neuroleptic malignant syndrome. Symptoms occur particularly when patients are relaxed, especially when they are lying down or sitting, and they lead to an urge to move about. The cause is unknown, but the disorder is especially common among pregnant women and also among uremic or diabetic patients with neuropathy. In most patients, no obvious predisposing cause is found, but several genetic loci have been associated with it. Symptoms may resolve with correction of coexisting irondeficiency anemia and often respond to dopamine agonists, levodopa, diazepam, clonazepam, gabapentin, or opiates. Dopaminergic therapy is the preferred treatment for restless legs syndrome and should be initiated with long-acting dopamine agonists (eg, pramipexole 0. Augmentation refers to the earlier onset or enhancement of symptoms; earlier onset of symptoms at rest; and a briefer response to medication. If it occurs in patients receiving an agonist, the daily dose should be divided, another agonist tried, or other medications substituted. Dopamine agonist therapy may be associated with development of impulse control disorders.
Syndromes
- AIDS.gov - www.aids.gov
- Use the shower chair
- Beckwith-Wiedemann syndrome
- An intravenous line (IV) will be placed into one of your veins. Medicines and fluids pass through this IV.
- Inflammation (pericarditis) or fluid in the sac around the heart (pericardial effusion)
- Partial webbing or fusing of fingers or toes
Cheap florinef online
Food decreases the peak plasma concentration but not the area under the concentration curve (see Chapter 3) gastritis diet kits order generic florinef. Hepatic impairment causes a slight decrease in clearance and may necessitate a lower dose. The agents described in the previous section are effective for the treatment of focal onset seizures, including focal-to-bilateral tonicclonic seizures (secondarily generalized tonic-clonic seizures), but some can worsen certain seizure types in generalized epilepsy syndromes. In addition, clinical trials of lamotrigine have demonstrated effectiveness in the treatment of generalized tonicclonic seizures (in idiopathic generalized epilepsy) and in the treatment of generalized absence epilepsy. The drug is generally well tolerated; however, it can produce a potentially fatal rash (StevensJohnson syndrome). Although adverse effects are similar to those of other sodium channel-blocking antiseizure drugs, lamotrigine paradoxically may cause insomnia instead of sedation. Lamotrigine causes fewer adverse cognitive effects than carbamazepine or topiramate. It can also improve depression in patients with epilepsy and reduces the risk of relapse in bipolar disorder. Lamotrigine is also approved for primary generalized tonic-clonic seizures and generalized seizures of the Lennox-Gastaut syndrome. Adverse effects include dizziness, headache, diplopia, nausea, insomnia, somnolence, and skin rash. Although the risk of rash may be diminished by introducing the drug slowly, pediatric patients are at greater risk. The drug has linear kinetics and is metabolized primarily by glucuronidation in the liver to the inactive 2-N-glucuronide, which is excreted in the urine. Lamotrigine is effective in the treatment of focal seizures in adults at dosages typically between 100 and 300 mg/d. Therapeutic serum levels have not been established, but toxicity is infrequent with levels < 10 mcg/mL. The combination of lamotrigine and valproate is believed to be particularly efficacious. However, valproate causes a two-fold increase in the half-life of lamotrigine and can increase blood levels correspondingly, leading to a risk of skin rash if valproate is added to a stable regimen of lamotrigine. In patients receiving valproate, the initial dose of lamotrigine must be reduced to 12. Chemistry Lamotrigine was developed when investigators thought that the antifolate effects of certain antiseizure drugs such as phenytoin might contribute to their effectiveness. Several phenyltriazines were developed; although their antifolate properties were weak, some were active in seizure screening tests. The antifolate activity of lamotrigine is not believed to contribute to its therapeutic activity in epilepsy. H2N N Lamotrigine Mechanism of Action the action of lamotrigine on voltage-gated sodium channels is similar to that of carbamazepine. The mechanism by which lamotrigine is effective against absence seizures is not known. Clinical Uses Although most controlled studies have evaluated lamotrigine as add-on therapy, the drug is effective as monotherapy for focal seizures, and lamotrigine is now widely prescribed for this indication because of its excellent tolerability. The drug accesses the luminal side of recycling synaptic vesicles by vesicular endocytosis. Clinical Uses Levetiracetam is effective in the treatment of focal seizures in adults and children, primary generalized tonic-clonic seizures, and the myoclonic seizures of juvenile myoclonic epilepsy. Adverse effects include somnolence, asthenia, ataxia, infection (colds), and dizziness. Less common but more serious are behavioral and mood changes, such as irritability, aggression, agitation, anger, anxiety, apathy, depression, and emotional lability. Oral formulations include extended-release tablets; an intravenous preparation is also available. Coadministration of brivaracetam with carbamazepine may increase exposure to carbamazepine epoxide, the active metabolite of carbamazepine, possibly leading to adverse effects; carbamazepine dose reduction should be considered. Similarly, coadministration of brivaracetam with phenytoin may increase phenytoin levels. Coadministration of other antiseizure drugs is unlikely to affect brivaracetam exposure. Two-thirds of the drug is excreted unchanged in the urine and the remainder as the inactive deaminated metabolite 2-pyrrolidone-N-butyric acid. In generalized convulsive seizures, whether occurring as a secondarily generalized convulsion following a focal seizure or as a primary generalized seizure, excitatory cortical neurons engage subcortical centers, including the thalamus, that relay the excitation throughout both hemispheres. Perampanel is therefore well suited to inhibit this spread of excitation, which may account for its activity in preventing secondary and primary generalized convulsive seizures. Perampanel binds to an allosteric site on the extracellular side of the channel, acting as a wedge to prevent channel opening. Whether it will prove to have the broadspectrum activity of levetiracetam remains to be demonstrated; given the similarity of the mechanisms of action, however, a broad spectrum is expected. Perampanel use is often associated with behavioral adverse reactions including aggression, hostility, irritability, and anger. The frequency of these adverse effects increases in a dosedependent fashion, and they occur more often in younger patients and in those with learning disabilities or dementia. Chemistry Four barbituric acid derivatives were once used for epilepsy: phenobarbital, mephobarbital, metharbital, and primidone. Pharmacokinetics Perampanel has a long half-life, typically ranging from 70 to 110 hours, which permits once-daily dosing. Clinical Uses Phenobarbital is useful in the treatment of focal seizures and generalized tonic-clonic seizures. Evidence-based comparisons of phenobarbital with phenytoin and carbamazepine have shown no difference in seizure control, but phenobarbital was more likely to be discontinued due to adverse effects. Phenobarbital may be useful in the treatment of myoclonic seizures, such as in juvenile myoclonic epilepsy, but it is not a drug of first choice. Long-term administration of phenobarbital leads to physical dependence such that seizure threshold is reduced upon withdrawal. The drug must be discontinued gradually over several weeks to avoid the occurrence of severe seizures or status epilepticus. Perampanel may decrease the effectiveness of levonorgestrel-containing hormonal contraceptives. Pharmacokinetics, Therapeutic Levels, & Dosage For pharmacokinetics, drug interactions, and toxicity of phenobarbital, see Chapter 22. The accepted serum concentration reference range is 15 to 40 mcg/mL, although many patients tolerate chronic levels above 40 mcg/mL.
Purchase florinef 0.1 mg online
The 1 blockers do not adversely and may even beneficially affect plasma lipid profiles gastritis diet ����� best 0.1 mg florinef, but this action has not been shown to confer any benefit on clinical outcomes. It has been available for many years, although it was initially thought not to be particularly effective because tachyphylaxis to its antihypertensive effects developed rapidly. The benefits of combination therapy are now recognized, and hydralazine may be used more effectively, particularly in severe hypertension. The combination of hydralazine with nitrates is effective in heart failure and should be considered in patients with both hypertension and heart failure, especially in African-American patients. Pharmacokinetics & Dosage Hydralazine is well absorbed and rapidly metabolized by the liver during the first pass, so that bioavailability is low (averaging 25%) and variable among individuals. As a consequence, rapid acetylators have greater first-pass metabolism, lower blood levels, and less antihypertensive benefit from a given dose than do slow acetylators. Even more than with hydralazine, the use of minoxidil is associated with reflex sympathetic stimulation and sodium and fluid retention. Toxicity Tachycardia, palpitations, angina, and edema are observed when doses of co-administered blockers and diuretics are inadequate. Headache, sweating, and hypertrichosis (the latter particularly bothersome in women) are relatively common. Topical minoxidil (as Rogaine) is used as a stimulant to hair growth for correction of baldness. The higher dosage was selected as the dose at which there is a small possibility of developing the lupus erythematosus-like syndrome described in the next section. However, higher dosages result in greater vasodilation and may be used if necessary. Nitroprusside dilates both arterial and venous vessels, resulting in reduced peripheral vascular resistance and venous return. The action occurs as a result of activation of guanylyl cyclase, either via release of nitric oxide or by direct stimulation of the enzyme. In the absence of heart failure, blood pressure decreases, owing to decreased vascular resistance, whereas cardiac output does not change or decreases slightly. In patients with heart failure and low cardiac output, output often increases owing to afterload reduction. In patients with ischemic heart disease, reflex tachycardia and sympathetic stimulation may provoke angina or ischemic arrhythmias. The syndrome is not associated with renal damage and is reversed by discontinuance of hydralazine. Peripheral neuropathy and drug fever are other serious but uncommon adverse effects. The effect results from the opening of potassium channels in smooth muscle membranes by minoxidil sulfate, the active metabolite. Increased potassium permeability stabilizes the membrane at its resting potential and makes contraction less likely. Because of its greater potential antihypertensive effect, minoxidil should replace hydralazine when maximal doses of the latter are not effective or in patients with renal failure and severe hypertension, who do not respond well to hydralazine. It is rapidly metabolized by uptake into red blood cells with release of nitric oxide and cyanide. Cyanide in turn is metabolized by the mitochondrial enzyme rhodanese, in the presence of a sulfur donor, to the less toxic thiocyanate. Thiocyanate is distributed in extracellular fluid and slowly eliminated by the kidney. Higher rates of infusion, if continued for more than an hour, may result in toxicity. Because of its efficacy and rapid onset of effect, nitroprusside should be administered by infusion pump and arterial blood pressure continuously monitored via intra-arterial recording. Toxicity Other than excessive blood pressure lowering, the most serious toxicity is related to accumulation of cyanide; metabolic acidosis, arrhythmias, excessive hypotension, and death have resulted. In a few cases, toxicity after relatively low doses of nitroprusside suggested a defect in cyanide metabolism. Administration of sodium thiosulfate as a sulfur donor facilitates metabolism of cyanide. Both have been advocated for prophylaxis or treatment of cyanide poisoning during nitroprusside infusion. Thiocyanate may accumulate over the course of prolonged administration, usually several days or more, particularly in patients with renal insufficiency who do not excrete thiocyanate at a normal rate. Thiocyanate toxicity is manifested as weakness, disorientation, psychosis, muscle spasms, and convulsions, and the diagnosis is confirmed by finding serum concentrations greater than 10 mg/ dL. Rarely, delayed hypothyroidism occurs, owing to thiocyanate inhibition of iodide uptake by the thyroid. Its halflife is approximately 24 hours, but the relationship between blood concentration and hypotensive action is not well established. When diazoxide was first marketed for use in hypertension, a dose of 300 mg by rapid injection was recommended. Because of reduced protein binding, smaller doses should be administered to persons with chronic renal failure. The hypotensive effects of diazoxide are also greater when patients are pretreated with blockers to prevent the reflex tachycardia and associated increase in cardiac output. Toxicity the most significant toxicity from parenteral diazoxide has been excessive hypotension, resulting from the original recommendation to use a fixed dose of 300 mg in all patients. The reflex sympathetic response can provoke angina, electrocardiographic evidence of ischemia, and cardiac failure in patients with ischemic heart disease, and diazoxide should be avoided in this situation. Occasionally, hyperglycemia complicates diazoxide use, particularly in persons with renal insufficiency. In contrast to the structurally related thiazide diuretics, diazoxide causes renal salt and water retention. However, because the drug is used for short periods only, this is rarely a problem. Because of its arteriolar dilating property, it was formerly used parenterally to treat hypertensive emergencies. Injection of diazoxide results in a rapid fall in systemic vascular resistance and mean arterial blood pressure. Diazoxide inhibits insulin release from the pancreas (probably by opening potassium channels in the beta cell membrane) and is used to treat hypoglycemia secondary to insulinoma. It acts primarily as an agonist of dopamine D1 receptors, resulting in dilation of peripheral arteries and natriuresis. The commercial product is a racemic mixture with the (R)-isomer mediating the pharmacologic activity. As with other direct vasodilators, the major toxicities are reflex tachycardia, headache, and flushing. Fenoldopam also increases intraocular pressure and should be avoided in patients with glaucoma. Diazoxide is similar chemically to the thiazide diuretics but has no diuretic activity. Clevidipine is a newer member of this group that is formulated for intravenous use only. Hemodynamic differences among calcium channel blockers may influence the choice of a particular agent. Nifedipine and the other dihydropyridine agents are more selective as vasodilators and have less cardiac depressant effect than verapamil and diltiazem. Reflex sympathetic activation with slight tachycardia maintains or increases cardiac output in most patients given dihydropyridines. Verapamil has the greatest depressant effect on the heart and may decrease heart rate and cardiac output.
Generic florinef 0.1mg fast delivery
As a consequence gastritis diet ���� buy 0.1mg florinef free shipping, behavior becomes compulsive; that is, decisions are no longer planned and under control, but automatic, which is the hallmark of addiction. This appealing hypothesis has been challenged based on the observation that some reward and drug-related learning is still possible in the absence of dopamine. Only when transporters of other biogenic amines are also knocked out does cocaine completely lose its rewarding properties. When cocaine is given, these transporters are also inhibited and dopamine is again increased. The neurons that are activated by aversive stimuli preferentially project to the prefrontal cortex, while the dopamine neurons inhibited by aversive stimuli are those that mostly target the nucleus accumbens. Regardless of the many roles of dopamine under physiologic conditions, all addictive drugs significantly increase its concentration in target structures of the mesolimbic projection. This suggests that high levels of dopamine may actually be at the origin of the adaptive changes that underlie dependence and addiction, a concept that is now supported by novel techniques that allow controlling the activity of dopamine neurons in vivo. In other words, cravings may recur at the presentation of contextual cues (eg, people, places, or drug paraphernalia). Non-substance-dependent disorders, such as pathologic gambling and compulsive shopping, share many clinical features of addiction. Several lines of arguments suggest that they also share the underlying neurobiologic mechanisms. Other patients may develop a habit for recreational activities, such as shopping, eating compulsively, or hypersexuality. Although large-scale studies are not yet available, an estimated one in seven parkinsonian patients develops an addiction-like behavior when receiving dopamine agonists (see chapter 28). Large individual differences exist also in vulnerability to substance-related addiction. Whereas one person may become "hooked" after a few doses, others may be able to use a drug occasionally during their entire lives without ever having difficulty in stopping. Even when dependence is induced with chronic exposure, only a small percentage of dependent users progress to addiction. For example, a retrospective analysis shows that after several decades of cocaine abuse, only 20% become addicted. A similar percentage for cocaine is also observed in rats and mice that have extended access to the drug. Correlated pre- and postsynaptic activity durably enhances synaptic efficacy and triggers the formation of new connections. Manipulations in mice that prevent or reverse drug-evoked plasticity in vivo also have effects on persistent changes of drug-associated behavioral sensitization or cue-induced drug seeking, providing more direct evidence for a causal role of synaptic plasticity in drug-adaptive behavior. Together, a circuit model of staged drug-evoked synaptic plasticity is emerging, whereby various symptoms are caused by changes in specific projections, eventually combining into addiction. Recent studies in rats suggest that impulsivity or excessive anxiety may be crucial traits that represent a risk for addiction. The transition to addiction is determined by a combination of environmental and genetic factors. Heritability of addiction, as determined by comparing monozygotic with dizygotic twins, is relatively modest for cannabinoids but very high for cocaine. Further genomic analysis indicates that numerous, perhaps even hundreds of alleles need to function in combination to produce the phenotype. Although some substance-specific candidate genes have been identified (eg, alcohol dehydrogenase, nicotinic acetylcholine receptor subunits), future research will also focus on genes implicated in the neurobiologic mechanisms common to all addictive drugs. An appealing idea, now supported by experimental evidence, is the contribution of epigenetics as a determinant of addiction vulnerability. The cellular mechanism involved and the relationship to synaptic plasticity are currently under investigation. Unlike addictive drugs, which primarily target the mesolimbic dopamine system, these agents primarily target cortical and thalamic circuits. These excitatory afferents mainly come from the thalamus and carry sensory information of varied modalities, which may constitute a link to enhanced perception. High doses of dextromethorphan, an over-the-counter cough suppressant, can also elicit a dissociative state. Concurrent effects on both thalamocortical and mesolimbic systems also exist for other addictive drugs. Psychosis-like symptoms can be observed with cannabinoids, amphetamines, and cocaine, which may reflect their effects on thalamocortical structures. The second group includes nicotine, alcohol, the benzodiazepines, dissociative anesthetics, and some inhalants, which interact with ionotropic receptors or ion channels. The last group comprises cocaine, amphetamines, and ecstasy, which all bind to monoamine transporters. The withdrawal syndrome may be very severe (except for codeine) and includes intense dysphoria, nausea or vomiting, muscle aches, lacrimation, rhinorrhea, mydriasis, piloerection, sweating, diarrhea, yawning, and fever. Beyond the withdrawal syndrome, which usually lasts no longer than a few days, individuals who have received opioids as analgesics only rarely develop addiction. The relative risk of addiction is 4 out of 5 on a scale of 1 (nonaddictive) to 5 (highly addictive). Treatment the opioid antagonist naloxone reverses the effects of a dose of morphine or heroin within minutes. This may be life-saving in the case of a massive overdose (see Chapters 31 and 58). Naloxone administration also provokes an acute withdrawal (precipitated abstinence) syndrome in a dependent person who has recently taken an opioid. In the treatment of opioid addiction, a long-acting opioid (eg, methadone, buprenorphine, morphine sulphate) is often substituted for the shorter-acting, more rewarding, opioid (eg, heroin). For substitution therapy, methadone is given orally once daily, facilitating supervised intake. Using a partial agonist (buprenorphine) and the much longer half-life (methadone, morphine sulphate, and buprenorphine) may also have some beneficial effects (eg, weaker drug sensitization, which typically requires intermittent exposures), but it is important to realize that abrupt termination of methadone administration invariably precipitates a withdrawal syndrome; that is, the subject on substitution therapy remains dependent. Some countries (eg, Canada, Denmark, Netherlands, United Kingdom, Switzerland) even allow substitution of medical heroin for street heroin. A followup of a cohort of addicts who received heroin injections in a controlled setting and had access to counseling indicates that addicts under heroin substitution have an improved health status and are better integrated in society. Pharmacology & Clinical Aspects As described in Chapter 31, opioids comprise a large family of endogenous and exogenous agonists at three G protein-coupled receptors: the -, -, and -opioid receptors. Although all three receptors couple to inhibitory G proteins (ie, they all inhibit adenylyl cyclase), they have distinct, sometimes even opposing effects, mainly because of the cell type-specific expression throughout the brain. This may explain why -opioid agonists cause euphoria, whereas agonists induce dysphoria. In line with the latter observations, the rewarding effects of morphine are absent in knockout mice lacking receptors but persist when either of the other opioid receptors are ablated. The most commonly abused opioids include morphine, heroin (diacetylmorphine, which is rapidly metabolized to morphine), codeine, and oxycodone. Because of such backward signaling, endocannabinoids are called retrograde messengers. In the hippocampus, release of endocannabinoids from pyramidal neurons selectively affects inhibitory transmission and may contribute to the induction of synaptic plasticity during learning and memory formation. Users also report feelings of well-being, grandiosity, and altered perception of passage of time. Dose-dependent perceptual changes (eg, visual distortions), drowsiness, diminished coordination, and memory impairment may occur. Cannabinoids can also create a dysphoric state and, in rare cases following the use of very high doses, eg, in hashish, result in visual hallucinations, depersonalization, and frank psychotic episodes. Today, medical use of botanical marijuana has been legalized in 25 states and the District of Columbia. Nevertheless this continues to be a controversial issue, mainly because of the fear that cannabinoids may serve as a gateway to the consumption of "hard" drugs or cause schizophrenia in individuals with a predisposition.
Order florinef 0.1mg on-line
If the sodium current is blocked over a critical length of the nerve gastritis loose stools buy genuine florinef online, propagation across the blocked area is no longer possible. In myelinated nerves, the critical length appears to be two to three nodes of Ranvier. At the minimum dose required to block propagation, the resting potential is not significantly altered. As a result, the refractory period is lengthened and the nerve conducts fewer action potentials. Elevated extracellular calcium partially antagonizes the action of local anesthetics owing to the calcium-induced increase in the surface potential on the membrane (which favors the low-affinity rested state). Conversely, increases in extracellular potassium depolarize the membrane potential and favor the inactivated state, enhancing the effect of local anesthetics. A series of 25 pulses was applied, and the resulting sodium currents (downward deflections) are superimposed. Note that the current produced by the pulses rapidly decreased from the first to the 25th pulse. A long rest period after the train resulted in recovery from block, but the block could be reinstated by a subsequent train. Other effects-Currently used local anesthetics bind to the sodium channel with low affinity and poor specificity, and there are multiple other sites for which their affinity is nearly the same as that for sodium channel binding. The role that such ancillary effects play in achievement of local anesthesia appears to be important but is poorly understood. Further, interactions with these other sites are likely the basis for numerous differences between the local anesthetics with respect to anesthetic effects (eg, differential block) and toxicities that do not parallel anesthetic potency, and thus are not adequately accounted for solely by blockade of the voltage-gated sodium channel. The actions of circulating local anesthetics at such diverse sites exert a multitude of effects, some of which go beyond pain control, including some that are also potentially beneficial. For example, there is evidence to suggest that the blunting of the stress response and improvements in perioperative outcome that may occur with epidural anesthesia derive in part from an action of the anesthetic beyond its sodium channel block. Circulating anesthetics also demonstrate antithrombotic effects having an impact on coagulation, platelet aggregation, and the microcirculation, as well as modulation of inflammation. Structure-Activity Characteristics of Local Anesthetics the smaller and more highly lipophilic local anesthetics have a faster rate of interaction with the sodium channel receptor. The latter agents are more potent and have longer durations of local anesthetic action. These long-acting local anesthetics also bind more extensively to proteins and can be displaced from these binding sites by other protein-bound drugs. Differential block-Since local anesthetics are capable of blocking all nerves, their actions are not limited to the desired loss of sensation from sites of noxious (painful) stimuli. With central neuraxial techniques (spinal or epidural), motor paralysis may impair respiratory activity, and autonomic nerve blockade may promote hypotension. Further, while motor paralysis may be desirable during surgery, it may be a disadvantage in other settings. For example, motor weakness occurring as a consequence of epidural anesthesia during obstetrical labor may limit the ability of the patient to bear down (ie, "push") during delivery. Similarly, when used for postoperative analgesia, weakness may hamper ability to ambulate without assistance and pose a risk of falling, while residual autonomic blockade may interfere with bladder function, resulting in urinary retention and the need for bladder catheterization. These issues are particularly problematic in the setting of ambulatory (same-day) surgery, which represents an ever-increasing percentage of surgical caseloads. Intrinsic susceptibility of nerve fibers-Nerve fibers differ significantly in their susceptibility to local anesthetic blockade. It has been traditionally taught, and still often cited, that local anesthetics preferentially block smaller diameter fibers first because the distance over which such fibers can passively propagate an electrical impulse is shorter. However, a variable proportion of large fibers are blocked prior to the disappearance of the small fiber component of the compound action potential. Most notably, myelinated nerves tend to be blocked before unmyelinated nerves of the same diameter. Another important factor underlying differential block derives from the state- and use-dependent mechanism of action of local anesthetics. Sensory (pain) fibers have a high firing rate and relatively long action potential duration. As type A delta and C fibers participate in high-frequency pain transmission, this characteristic may favor blockade of these fibers earlier and with lower concentrations of local anesthetics. The potential impact of such effects mandates cautious interpretation of non-physiologic experiments evaluating intrinsic susceptibility of nerves to conduction block by local anesthetics. Anatomic arrangement-In addition to the effect of intrinsic vulnerability to local anesthetic block, the anatomic organization of the peripheral nerve bundle may impact the onset and susceptibility of its components. As one would predict based on the necessity of having proximal sensory fibers join the nerve trunk last, the core will contain sensory fibers innervating the most distal sites. Anesthetic placed outside the nerve bundle will thus reach and anesthetize the proximal fibers located at the outer portion of the bundle first, and sensory block will occur in sequence from proximal to distal. Fiber Type Type A Alpha Beta Gamma Delta Type B Type C Dorsal root Sympathetic Pain Postganglionic 0. When local anesthetics are injected extradurally, it is referred to as an epidural block. A caudal block is a specific type of epidural block in which a needle is inserted into the caudal canal via the sacral hiatus. Injections around peripheral nerves are known as perineural blocks (eg, paravertebral block). Finally, injection into cerebrospinal fluid in the subarachnoid (intrathecal) space is referred to as a spinal block. Clinical Block Characteristics In clinical practice, there is generally an orderly evolution of block components beginning with sympathetic transmission and progressing to temperature, pain, light touch, and finally motor block. This is most readily appreciated during onset of spinal anesthesia, where a spatial discrepancy can be detected in modalities, the most vulnerable components achieving greater dermatomal (cephalad) spread. Thus, loss of the sensation of cold (often assessed by a wet alcohol sponge) will be roughly two segments above the analgesic level for pinprick, which in turn will be roughly two segments rostral to loss of light touch recognition. However, because of the anatomic considerations noted earlier for peripheral nerve trunks, onset with peripheral blocks is more variable, and proximal motor weakness may precede onset of more distal sensory loss. Additionally, anesthetic solution is not generally deposited evenly around a nerve bundle, and longitudinal spread and radial penetration into the nerve trunk are far from uniform. With respect to differential block, it is worth noting that "successful" surgical anesthesia may require loss of touch, not just ablation of pain, as some patients will find even the sensation of touch distressing during surgery, often fearing that the procedure may become painful. Effect of Added Vasoconstrictors Several benefits may be derived from addition of a vasoconstrictor to a local anesthetic. First, localized neuronal uptake is enhanced because of higher sustained local tissue concentrations that can translate clinically into a longer duration block. This may enable adequate anesthesia for more prolonged procedures, extended duration of postoperative pain control, and lower total anesthetic requirement. Second, peak blood levels will be lowered as absorption is more closely matched to metabolism and elimination, and the risk of systemic toxic effects is reduced. Moreover, when incorporated into a spinal anesthetic, epinephrine may not only contribute to prolongation of the local anesthetic effect via its vasoconstrictor properties, but also exert a direct analgesic effect mediated by postsynaptic 2 adrenoceptors within the spinal cord. Recognition of this potential has led to the clinical use of the 2 agonist clonidine as a local anesthetic adjuvant for spinal anesthesia. The addition of epinephrine to anesthetic solutions can potentiate the neurotoxicity of local anesthetics used for peripheral nerve blocks or spinal anesthesia. Further, the use of a vasoconstrictor agent in an area that lacks adequate collateral flow (eg, digital block) is generally avoided, although some have questioned the validity of this proscription. Intentional Use of Systemic Local Anesthetics Although the principal use of local anesthetics is to achieve anesthesia in a restricted area, these agents are sometimes deliberately administered systemically to take advantage of suppressive effects on pain processing. In addition to documented reductions in anesthetic requirement and postoperative pain, systemic administration of local anesthetics has been used with some success in the treatment of chronic pain, and this effect may outlast the duration of anesthetic exposure. The achievement of pain control by systemic administration of local anesthetics is thought to derive, at least in part, from the suppression of abnormal ectopic discharge, an effect observed at concentrations of local anesthetic an order of magnitude lower than those required for blockade of propagation of action potentials in normal nerves. Consequently, these effects can be achieved without the adverse effects that would derive from failure of normal nerve conduction. Escalating doses of anesthetic appear to exert the following systemic actions: (1) low concentrations may preferentially suppress ectopic impulse generation in chronically injured peripheral nerves; (2) moderate concentrations may suppress central sensitization, which would explain therapeutic benefit that may extend beyond the anesthetic exposure; and (3) higher concentrations will produce general analgesic effects and may culminate in serious toxicity. Systemic Toxicity the dose of local anesthetic used for epidural anesthesia or high-volume peripheral blocks is sufficient to produce major clinical toxicity, even death.
Cheap 0.1mg florinef visa
These doses are used to calculate the initial dose to be tried in humans gastritis diet ������ order 0.1 mg florinef with mastercard, usually taken as one hundredth to one tenth of the noeffect dose in animals. Two to 6 years may be required to collect and analyze data on toxicity before the drug can be considered ready for testing in humans. Scientists are properly concerned about this situation, and progress has been made toward reducing the numbers required while still obtaining valid data. Cell and tissue culture in vitro methods and computer modeling are increasingly being used, but their predictive value is still limited. Nevertheless, some segments of the public attempt to halt all animal testing in the unfounded belief that it has become unnecessary. Extrapolations of toxicity data from animals to humans are reasonably predictive for many but not for all toxicities. For statistical reasons, rare adverse effects are unlikely to be detected in preclinical testing. Candidate drugs that survive the initial screening procedures must be carefully evaluated for potential risks before and during clinical testing. Type of Test Acute toxicity Subacute or subchronic toxicity Approach and Goals Usually two species, two routes. In some cases, determine the acute dose that is lethal in approximately 50% of animals. Test effects on animal mating behavior, reproduction, parturition, progeny, birth defects, postnatal development. Test effects on genetic stability and mutations in bacteria (Ames test) or mammalian cells in culture; dominant lethal test and clastogenicity in mice. Scientifically valid results are not guaranteed simply by conforming to government regulations, however, and the design and execution of a good clinical trial require interdisciplinary personnel including basic scientists, clinical pharmacologists, clinician specialists, statisticians, and others. The need for careful design and execution is based on three major confounding factors inherent in the study of any drug in humans. There is growing interest in analyzing genetic variations as part of the trial that may influence whether a person responds to a particular drug. It has been shown that age, gender, and pregnancy influence the pharmacokinetics of some drugs, but these factors have not been adequately studied because of legal restrictions and reluctance to expose these populations to unknown risks. Subject and Observer Bias and Other Factors Most patients tend to respond in a positive way to any therapeutic intervention by interested, caring, and enthusiastic medical personnel. The manifestation of this phenomenon in the subject is the placebo response (Latin, "I shall please") and may involve objective physiologic and biochemical changes as well as changes in subjective complaints associated with the disease. The placebo response is usually quantitated by administration of an inert material with exactly the same physical appearance, odor, consistency, etc, as the active dosage form. The magnitude of the response varies considerably from patient to patient and may also be influenced by the duration of the study. Placebo adverse effects and "toxicity" also occur but usually involve subjective effects: stomach upset, insomnia, sedation, and so on. Subject bias effects can be quantitated-and minimized relative to the response measured during active therapy-by the single-blind design. This involves use of a placebo as described above, administered to the same subjects in a crossover design, if possible, or to a separate control group of well-matched subjects. In this design, a third party holds the code identifying each medication packet, and the code is not broken until all the clinical data have been collected. Drug effects seen in clinical trials are obviously affected by the patient taking the drugs at the dose and frequency prescribed. In a recent phase 2 study, one third of the patients who said they were taking the drug were found by blood analysis to have not taken the drug. Confirmation of compliance with protocols (also known as adherence) is a necessary element to consider. The various types of studies and the conclusions that may be drawn from them are described in the accompanying text box. The Variable Natural History of Most Diseases Many diseases tend to wax and wane in severity; some disappear spontaneously, even, on occasion, cancer. A good experimental design takes into account the natural history of the disease by evaluating a large enough population of subjects over a sufficient period of time. Further protection against errors of interpretation caused by disease fluctuations is sometimes provided by using a crossover design, which consists of alternating periods of administration of test drug, placebo preparation (the control), and the standard treatment (positive control), if any, in each subject. These sequences are systematically varied, so that different subsets of patients receive each of the possible sequences of treatment. The Presence of Other Diseases and Risk Factors Known and unknown diseases and risk factors (including lifestyles of subjects) may influence the results of a clinical study. For example, some diseases alter the pharmacokinetics of drugs (see Chapters 3 through 5). Concentrations of blood or tissue components being monitored as a measure of the effect of the new agent may be influenced by other diseases or other drugs. Attempts to avoid this hazard usually involve the crossover technique (when feasible) and proper selection and assignment of patients to each of the study groups. This requires obtaining accurate diagnostic tests and medical and pharmacologic histories (including use of recreational drugs, over-the-counter drugs, and "supplements") and the use of statistically valid methods of Drug Studies-The Types of Evidence* As described in this chapter, drugs are studied in a variety of ways, from 30-minute test tube experiments with isolated enzymes and receptors to decades-long observations of populations of patients. The conclusions that can be drawn from such different types of studies can be summarized as follows. Basic research is designed to answer specific, usually single, questions under tightly controlled laboratory conditions, eg, does drug x inhibit enzyme y The basic question may then be extended, eg, if drug x inhibits enzyme y, what is the concentration-response relationship Analytic epidemiologic studies consist of observations designed to test a specified hypothesis, eg, that thiazolidinedione antidiabetic drugs are associated with adverse cardiovascular events. Cohort epidemiologic studies utilize populations of patients that have (exposed group) and have not (control group) been exposed to the agents under study and ask whether the exposed groups show a higher or lower incidence of the effect. Case-control epidemiologic studies utilize populations of patients that have displayed the end point under study and ask whether they have been exposed or not exposed to the drugs in question. Such epidemiologic studies add weight to conjectures but cannot control all confounding variables and therefore cannot conclusively prove cause and effect. While the numbers may be dramatically increased by meta-analysis, the individual studies still suffer from their varying methods and end points, and a meta-analysis cannot prove cause and effect. Randomization is the best method for distributing all foreseen confounding factors, as well as unknown confounders, equally between the experimental and control groups. When properly carried out, such studies are rarely invalidated and are considered the gold standard in evaluating drugs. A critical factor in evaluating the data regarding a new drug is access to all the data. Unfortunately, many large studies are never published because the results are negative, ie, the new drug is not better than the standard therapy. This missing data phenomenon falsely exaggerates the benefits of new drugs because negative results are hidden. Shared responsibility results in complications when questions arise regarding the use of drugs, eg, antibiotics, in food animals. A different type of problem arises when so-called food supplements are found to contain active drugs, eg, sildenafil analogs in "energy food" supplements. If a drug has not been shown through adequately controlled testing to be "safe and effective" for a specific use, it cannot be marketed in interstate commerce for this use. For example, the Federal Food, Drug, and Cosmetic Act of 1938 was largely a reaction to deaths associated with the use of a preparation of sulfanilamide marketed before it and its vehicle were adequately tested. Similarly, the Kefauver-Harris Amendments of 1962 were, in part, the result of a teratogenic drug disaster involving thalidomide.
