

Proven xeloda 500mg
A common example of this would be decisions about not attempting cardiopulmonary resuscitation women's health center brooklyn order xeloda 500mg mastercard. A patient with a major stroke, who is unable to swallow but is expected to survive the event, will develop renal impairment and thirst if not given fluids and should be hydrated. On the other hand, when a patient has been deteriorating and is clearly dying, parenteral hydration needs very careful consideration and it is very important to manage this on an individual basis. Where a patient and family are happy with meticulous oral hygiene and care to reduce the sensation of dryness in the mouth, this is usually more appropriate and effective at the end of life than parenteral hydration, which by itself will not necessarily improve the sensation of dryness. In some patients, parenteral hydration will simply exacerbate pooling of secretions, causing noisy and distressing breathing. As in other palliative situations, it may not be possible to identify and treat the underlying cause, and the focus of management should be to ensure that the patient is comfortable. It is important to distinguish between behavioural change due to pain and that due to delirium, as opioids will improve one and worsen the other. It is important, even in the care of the actively dying patient, to treat delirium with antipsychotic medicines, such as haloperidol, rather than to regard it as distress or anxiety and use benzodiazepines only. The overwhelming force in caring for any patient must be to listen to that patient and family and take their wishes on board. Patients know when health-care professionals are just receiving the information, as opposed to receiving and understanding the information in the context of the patient, their illness and needs, their carers and the socioeconomic context. It is impossible to provide holistic care for a patient without this comprehension. Every patient is unique and it is important to avoid slipping into a tick-box mentality in addressing items that should be covered in patients with advanced, incurable disease. While the key to successful palliative care is effective interdisciplinary working, every patient needs to know who has overall responsibility oo oo the wishes of the patient are paramount in Western societies, whereas in other cultures the views of the family are equally important. In order to decide what the patient would have wished, as much information as possible should be gained about any previously expressed wishes, along with the views of relatives and other health professionals. Once the conclusion has been reached that a patient is going to die in days to a few weeks, there is a significant shift in management (Box 34. Symptom control, relief of distress and care for the family become the most important elements of care. Medication and investigation are justifiable only if they contribute to these ends. When patients can no longer drink because they are dying, intravenous fluids are usually not necessary and may cause worsening of bronchial secretions; however, this is a decision that can be made only on an individual basis. Management should not be changed without discussion with the patient and/or family. For example, morphine or diamorphine may be used to control pain, levomepromazine to control nausea, haloperidol to treat delirium, diazepam or midazolam to treat distress, and hyoscine hydrobromide to reduce respiratory secretions. Side-effects, such as drowsiness, may be acceptable if the principal aim of relieving distress is achieved. Poor communication with families at this time is one of the most common reasons for family distress afterwards and for formal complaints. Doctors are sometimes poor at recognising this and should be alert to the views of other members of the multidisciplinary team. Families also are grateful for the chance to prepare themselves for the death of a relative, by timely and gentle discussion with the doctor or other health professionals. There are two important caveats: firstly, wishes can and do change as the terminal situation evolves, and secondly, planning in general can only be done over time as patients form a relationship with professionals and evolve an understanding of the situation in which they find themselves. Structures for assessment and planning around end-of-life care are for guidance only and the focus should evolve with the individual patient. Families and other carers are often unprepared for the challenge of caring for a dying person. It can be an exhausting experience, both emotionally and physically, and without a critical number of carers battle fatigue can ensue, resulting in urgent admissions. With much discussion about advance directives, we should not lose sight of the reality of changing circumstances and wishes. Good anticipatory care means not just providing for new physical symptoms, but also planning for any time when care at home becomes no longer possible. Many of these requests are transient; some are associated with poor control of physical symptoms or a depressive illness. All expressions of a wish to die are an opportunity to help the patient discuss and address unresolved issues and problems. Sometimes, patients may choose to discontinue life-prolonging treatments, such as diuretics or anticoagulation, following discussion and the provision of adequate alternative symptom control. However, there remain a small number of patients who have a sustained, competent wish to end their lives, despite good control of physical symptoms. Euthanasia is now permitted or legal under certain circumstances in some countries but remains illegal in many others; public, ethical and legal debate over this issue is likely to continue and is often influenced by many complex non-palliative care issues. The European Association for Palliative Care does not see euthanasia or physician-assisted suicide as part of the role of palliative care physicians. A comprehensive, high-quality review of the current evidence for the pharmacological management of neuropathic pain. For convenience, however, the litre (L) is used as the unit of volume in laboratory work. Many reference ranges vary between laboratories, depending on the assay method used and on other factors; this is especially the case for enzyme assays. No details are given here of the collection requirements, which may be critical to obtaining a meaningful result. Unless otherwise stated, reference ranges shown apply to adults; values in children may be different. Many analytes can be measured in either serum (the supernatant of clotted blood) or plasma (the supernatant of anticoagulated blood). An example is fibrinogen, where plasma is required, since fibrinogen is largely absent from serum. In contrast, serum is required for electrophoresis to detect paraproteins because fibrinogen migrates as a discrete band in the zone of interest. A number of hormones are unstable and collection details are critical to obtaining a meaningful result. Values in the table are only a guideline; hormone levels can often be meaningfully understood only in relation to factors such as gender, age, time of day, pubertal status, stage of the menstrual cycle, pregnancy and menopausal status. Reference ranges are usually dependent on the method used for analysis and frequently differ between laboratories. Hospital laboratories may provide reference ranges that are ageadjusted or based on pubertal stage but this is not always the case. It is therefore important for the doctor requesting these tests to understand the impact of age and puberty on interpretation of the results. Reference ranges for hormone results are described according to the Tanner stages of puberty (p. Precise identification of genes and their pathogenic mutations has injected an urgency among care providers to become aware of the rapidly escalating opportunities parents have to avoid having offspring with serious or fatal genetic disorders. For any health or life-threatening genetic disorder, prenatal diagnosis (or even preimplantation genetic diagnosis) has become a viable option, and should be offered. Even adult-onset malignant, neurodegenerative, cardiovascular and other serious systemic disorders now feature in the indications, not only for presymptomatic or predictive diagnosis, but for prenatal diagnosis. Given the wide scope of clinical genetics in all medical specialties, the need for clinicians to confer and refer has never been greater. The coalescence of advances in molecular genetics, fetal imaging and noninvasive prenatal screening, has culminated in the provision of new opportunities for the prevention or avoidance of genetic disorders and congenital malformations. In context, women at risk for having progeny with abnormalities expect to be informed about their odds and options, optimally during preconception counseling. Their concerns are serious, given the significant contribution of genetic disorders to morbidity and mortality in children and adults.
Syndromes
- Control risk factors, such as irregular heartbeat, high blood pressure, and high cholesterol
- What other symptoms are also present?
- Practice good oral hygiene. Brush your gums well to reduce the risk of getting another infection.
- Kidneys that are not working well and are becoming worse.
- The genitals and the skin around them lose skin color.
- Potatoes that have been baked, boiled, or mashed
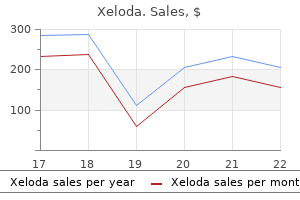
Order xeloda with american express
The distribution of pain can be documented on a diagram of the body menopause goddess buy discount xeloda 500mg on-line, on which the patient can mark the sites that are painful. Similarly, other methods have been developed with which to assess the severity of pain using verbal, numerical and behavioural rating scales. Visual scoring systems employing different facial expressions may be of value in paediatric patients and those with cognitive impairment. Documenting changes in pain scores using questionnaires can be helpful in indicating to what extent drug treatments have been successful and can reduce the time taken to achieve pain control. In those with focal pain, neurological examination should be performed, focusing particularly on any areas of abnormal sensation, reflexes and evidence of muscle wasting. A general examination should be carried out to determine whether there is any evidence of an underlying physical disorder that can account for the pain. In addition to the use of investigations to find the underlying cause of pain, patients with persistent or chronic pain may benefit from sensory testing or diagnostic nerve blocks to explore the underlying mechanisms and direct treatment. For example, a combined femoral and sciatic nerve block may be used in a patient with lower limb amputation to assess whether the pain is predominantly peripherally or centrally generated. If the pain is not improved by an effective nerve block, then peripherally directed therapies are unlikely to be effective. Nerve blockade can also be used to determine whether more radical therapies, such as nerve ablation, might be helpful in controlling pain, particularly that related to cancer. The past medical and medication history should be recorded and specific enquiry made about substance misuse and any previous history of physical or mental abuse. There are some patient populations in whom particular challenges arise, often related to differences in communication ability. Strategies that can be used to overcome these difficulties are summarised in Box 34. Several types of exercise have been successfully used delivered in various ways, through physiotherapists, exercise classes or individual tuition. In choosing a form of exercise therapy, it is important to tailor the approach most likely to be acceptable co. Self-management works best if the patient has some understanding of their chronic pain, and acceptance that it is unlikely to resolve completely. The aim is for patients to maximise their quality of life and function despite ongoing pain. Support for self-management can be delivered by health-care professionals, patients who suffer from the same condition or lay people, either on an individual basis, in a group setting or, increasingly, through web-based resources. There is a strong educational component to supported self-management, which seeks to generate an interaction between patient and tutor. In general, it is advisable to use a multimodal approach in the treatment of chronic pain, choosing different drugs to target pain processing at multiple points (Box 34. By employing different classes of analgesic, it is possible to use lower doses of each, thereby improving the side-effect profile. There is considerable inter-individual variability in response to analgesics, even within the same class. There are many reasons for this, including genetic variations in the enzymes that metabolise drugs. Examples include the use of immunosuppressive medication in inflammatory disease, chemotherapy, radiotherapy or hormone therapy in cancer, and antimicrobial therapy in patients with infection. There are many circumstances, however, in which the underlying cause of pain cannot be treated or the treatments available are incompletely effective. In all cases, a multidisciplinary approach is necessary that combines pharmacological management with supported self-management, and other specific interventions when appropriate. Because of this it is important to pace physical activity to ensure that patients do not cycle from over-activity, with a flare in pain, to fatigue and deconditioning. This can be done by working with patients to establish their baseline level of activity and using an individually tailored, graded exercise programme (Box 34. This may include normal household activities, as well as targeted exercises and stretches. Manual therapy covers a variety of hands-on treatments, including manipulation, mobilisation and massage. Manual therapy can be provided by a range of therapists, including physiotherapists, osteopaths and chiropractors. There is some evidence of shortterm benefit for manual therapy but limited evidence of long-term efficacy. There is also some of evidence that it activates inhibitory descending spinal pathways, via a serotonergic mechanism. Other postulated mechanisms include endocannabinoid re-uptake inhibition, and inhibition of nitric oxide and tumour necrosis factor alpha. For migraine and tension-type headache it has moderate efficacy at a dose of 1000 mg. It is used widely for musculoskeletal disorders and osteoarthritis, with very little high-quality evidence that it is much better than placebo, even at doses of up to 4000 mg per day. Acute liver failure is a well-recognised complication of paracetamol overdose but this risk may also be increased with long-term use, even within the recommended dose range. This is particularly important if metabolites are active, as is the case with codeine and tramadol, which are metabolised to morphine. Genetic variations have also been described in the opioid receptors and downstream pathways that they affect, with good pre-clinical evidence that variations in mu opioid receptors alter analgesic response to different opioids. Because of this there is a good rationale to try different drugs, even ones from the same class, if there is an inadequate response or there are unacceptable side-effects with one agent. Whatever drug or combination of drugs is chosen, the key to successful pharmacological management is careful assessment and review, aiming for an acceptable balance between the benefits of treatment in providing pain relief, maximising function, and improving quality of life and adverse effects. These drugs can be given systemically or locally and are discussed in more detail on page 1002. They are also useful in the management of pain in cancer patients, as discussed later in this chapter (p. Although widely prescribed, there is limited high-quality evidence of long-term efficacy in chronic pain, with a need for further studies in this area. A single application (done by a trained health-care professional) of a high-dose 8% capsaicin patch can give around 12 weeks of pain relief for neuropathic pain and can be repeated thereafter. Capsaicin activates the channel, causing an initial sensation of heat, but an analgesic effect subsequently results due to desensitisation of the channel. The mode of action is blockade of sodium channels in primary afferent neurons and nociceptors, which reduces peripheral input to the spinal cord. They are of particular value when used in combination in the management of pain with a neuropathic component but require careful dose titration over a number of weeks, to reach a dose that balances efficacy with side-effects. While the response to individual agents is variable, it is often possible to find an agent or combination of agents that works for most patients. Opioids are traditionally divided into subclasses of weak opioids, such as codeine and dihydrocodeine, and strong opioids, such as morphine and oxycodone. While tramadol is a weak agonist at the mu opioid receptor, it is classified as a strong opioid in some countries. The dosages and characteristics of commonly prescribed opioids are shown in Box 34. There is evidence of short- to medium-term benefit for strong opioids in low back pain and osteoarthritis but there have been very few good-quality studies of long-term use. Additionally, there is increasing concern about potential harm from long-term use. This includes addiction, dependence, opioid-induced hyperalgesia, endocrine dysfunction, fracture risk (especially in the elderly), overdose and cardiovascular re ks f ok s ks fre.
Buy xeloda 500mg cheap
Subsequently menstruation cramps relief purchase xeloda with mastercard, the lesion may become palpable and this is indicative of a vertical growth phase, with dermal invasion; when this occurs, the tumour has the potential to invade lymphatics and vessels and to become metastatic. The mitotic rate and the presence or absence of any evidence of lymphovascular or perineural involvement should also be ascertained. The clinical staging of melanoma extent is essential, in order to establish whether disease is primary and localised, or if there is nodal or metastatic spread. Wide excision of melanoma with a low risk of metastasis (stage 1 disease, Breslow thickness < 1 mm) with a 1 cm clear margin is accepted practice. For tumours with a Breslow thickness of 1 mm or more, a sentinel lymph node biopsy should be considered. This procedure provides additional prognostic information but there is no evidence that it improves survival. Local recurrence of disease and palpable local node involvement should be treated surgically. Localised cutaneous metastases or in transit disease may be amenable to palliation with electrochemotherapy if there is no evidence of widespread metastatic disease. Despite the major advances in treatment options for advanced melanoma, the prognosis for metastatic disease remains poor and treatment options are palliative. Genetic developments have facilitated the introduction of tumour-targeted treatments for advanced, unresectable and/or metastatic disease, such as the B-Raf and c-Kit kinase inhibitors for patients expressing these gene mutations, notably dabrafenib and vemurafenib, with demonstrable clinical responses. Standard chemotherapy may also be used in some cases of metastatic disease, although outcomes are poor. It is important for patients with advanced melanoma to be managed through a multidisciplinary team in order to optimise care and facilitate their inclusion in clinical trials. All patients should be advised regarding ongoing photoprotection, with sensible behaviour in the sun, covering up, wearing hats and high-factor sunscreen use. However, evidence has shown that despite patients with melanoma being advised to photoprotect, many follow this advice only for the first year following diagnosis, thus emphasising the need for ongoing reinforcement of guidance with regard to photoprotection. It is also prudent to advise patients who are photoprotecting to optimise oral vitamin D through diet and/or supplements. Survival rates fall to less than 10% for those with advanced nodal or metastatic disease. It is thought to be associated with chronic sun exposure and most commonly occurs on the central face. The classical appearance is of an isolated dome-shaped nodule often of 5 cm or more in diameter, with a central keratin plug. They are most common on sun-exposed sites in fair-skinned individuals, particularly children and those with red hair, and on the face. It is thought that they may arise as the result of abnormalities in the normal migration of melanocytes during development. Most melanocytic naevi appear in childhood and early adult life, or during pregnancy or oestrogen therapy. They occur in both sexes and with increasing age, and are most common on the face and trunk. If there is no doubt about the diagnosis, they can be left alone or treated by cryotherapy or curettage if they are cosmetically troublesome. If there is a suspicion of melanoma, excision or diagnostic biopsy should be undertaken. Benign melanocytic naevi and basal cell papillomas, in particular, can often be mistaken for melanoma, even by dermatologists. They can sometimes be difficult to distinguish from melanocytic lesions, particularly if they are thrombosed or occur on particular sites, such as the lip or genitalia. Lipoma Lipomas are benign tumours of adipocytes that are characteristically soft and lie more deeply in the skin than epidermal tumours; they are usually diagnosed easily on clinical grounds. Treatment is not required unless there is diagnostic doubt or they are symptomatic or cosmetically troublesome, in which case a diagnostic biopsy or surgical excision may be required. Compound and intradermal naevi are nodules because of the dermal component, and may be hair-bearing. Such naevi are known to occur in some rare families with an inherited melanoma predisposition. However, the significance of such changes in non-familial cases is unclear and there is no consensus on management and follow-up. Although approximately 50% of melanomas arise in pre-existing naevi, most naevi do not become malignant; although a changing naevus must be taken seriously, most will not be melanomas. Malignant change is most likely in large congenital melanocytic naevi (risk may correlate with the size of the lesion) and possibly in families who have been diagnosed as showing large numbers of atypical naevi with a history of melanoma. Treatment is not required unless there is diagnostic doubt or they are causing symptoms, such as irritation, or for cosmetic reasons. Melanocytic naevi are normal and do not require excision, unless malignancy is suspected or they become repeatedly inflamed or traumatised. Advice on photoprotection is important for fair-skinned individuals with multiple naevi. There are two main presentations: bullous impetigo, caused by a staphylococcal epidermolytic toxin, and non-bullous impetigo. All ages can be affected but non-bullous disease particularly affects young children, often in late summer. Its aetiology is unclear, although a reactive process secondary to insect bites or trauma is one hypothesis. There is frequently a ring of pigment around the lesion and dimpling when the skin is pinched, reflecting epidermal tethering. They may be difficult to distinguish from nodular melanoma and are therefore often excised. It is caused by staphylococcal toxins and early cases were thought to arise with tampon use. Staphylococcal folliculitis is most common in children and often occurs on the scalp or limbs. The focus of infection may be minor skin trauma, the umbilicus, urinary tract or nasopharynx. Systemic antibiotics and intensive supportive measures should be commenced immediately. Bacterial swabs from nostrils, axillae and groins should be taken from family members to exclude staphylococcal carriage. Predisposing factors include poor hygiene, malnutrition and underlying skin disease, such as scabies. It is commonly seen in drug abusers, and minor trauma can predispose to lesion development. B the condition was rapidly diagnosed by examination of a frozen section of skin snip. Predisposing factors are minor skin abrasions and the existence of other skin conditions, such as infestations or eczema. In non-bullous impetigo, a thin-walled vesicle develops; it rapidly ruptures and is rarely seen intact. The face, scalp and limbs are commonly affected but other sites can also be involved, particularly if there are predisposing factors such as eczema. A bacterial swab should be taken from blister fluid or an active lesion before treatment commences. Around one-third of the population is a nasal carrier of Staphylococcus, so swabs from the nostrils should also be obtained. In mild, localised disease, topical treatment with mupirocin or fusidic acid is usually effective and limits the spread of infection. The use of topical antiseptics and soap and water to remove infected crusts is also helpful.
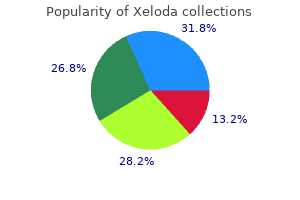
Cheap xeloda online visa
After being taken up by epithelial cells women's health stomach issues buy 500 mg xeloda overnight delivery, gluten peptides are deamidated by the enzyme tissue transglutaminase in the subepithelial layer. Endoscopic appearances should not preclude biopsy, as the mucosa usually looks normal. The histological features are usually characteristic but other causes of villous atrophy should be considered (Box 21. Sometimes the villi appear normal but there are excess numbers of intra-epithelial lymphocytes (lymphocytic duodenosis). In infancy, it occurs after weaning on to cereals and typically presents with diarrhoea, malabsorption and failure to thrive. In older children, it may present with non-specific features, such as delayed growth. Features of malnutrition are found on examination and mild abdominal distension may be present. Affected children have growth and pubertal delay, leading to short stature in adulthood. In adults, the disease usually presents during the third or fourth decade and females are affected twice as often as males. The presentation is highly variable, depending on the severity and extent of small bowel involvement. Some have florid malabsorption, while others develop non-specific symptoms, such as tiredness, weight loss, folate deficiency or iron deficiency anaemia. Unrecognised coeliac disease is associated with mild under-nutrition and osteoporosis. In some centres, people at higher risk of developing coeliac disease, such as those with type 1 diabetes, may undergo periodic antibody screening. Such screening may identify people with asymptomatic or minimally symptomatic disease; there is controversy about the optimum management strategy for such individuals. There is blunting of villi (B), crypt hyperplasia (H) and inflammatory infiltration of the lamina propria (I). If the antibody screen is positive, adult patients should remain on a gluten-containing diet until duodenal biopsies are taken. High-titre serology in children can be diagnostic without the need for endoscopy and biopsy. Anti-endomysial antibodies of the IgA class are detectable by immunofluorescence in most untreated cases. IgG antibodies, however, must be analysed in patients with coexisting IgA deficiency. Management re sf re sf sf re the aims are to correct existing deficiencies of micronutrients, such as iron, folate, calcium and/or vitamin D, and to achieve mucosal healing through a life-long gluten-free diet. Initially, frequent dietary counselling is required to make sure the diet is being observed, as the most common reason for failure to improve with dietary treatment is accidental or unrecognised gluten ingestion. Mineral and vitamin supplements are also given when indicated but are seldom necessary when a strict gluten-free diet is adhered to . Booklets produced by coeliac societies in many countries, containing diet sheets and recipes for the use of gluten-free flour, are of great value. There are currently no additional non-invasive tests to assess small bowel mucosal healing. Repeat small bowel biopsies are not required routinely but should be considered in patients whose symptoms fail to improve and those in whom antibody levels remain high. In these ks fre ks f ok s fre this is characterised by crops of intensely itchy blisters over the elbows, knees, back and buttocks (p. Immunofluorescence shows granular or linear IgA deposition at the dermo-epidermal junction. Almost all patients have partial villous atrophy on duodenal biopsy, identical to that seen in coeliac disease, even though they usually have no gastrointestinal symptoms. In contrast, fewer than 10% of coeliac patients have evidence of dermatitis herpetiformis, although both disorders are associated with the same histocompatibility antigen groups. The disease occurs mainly in the West Indies and in southern India, Malaysia and Indonesia. Although no single bacterium has been isolated, the condition often begins after an acute diarrhoeal illness. Small bowel bacterial overgrowth with Escherichia coli, Enterobacter and Klebsiella is frequently seen. Biochemical tests may reveal reduced concentrations of calcium, magnesium, total protein, albumin or vitamin D. Serum IgA measurement is required to ensure an appropriate IgA response and to allow analysis of serological testing. This diagnosis can be made by barium studies or enteroscopy but laparotomy and full-thickness biopsy may be required. Glucocorticoids are used with mixed success and some patients require surgical resection and parenteral nutrition. Osteoporosis and osteomalacia may occur in patients with longstanding, poorly controlled coeliac disease. These complications are less common in those who adhere strictly to a gluten-free diet. There remain a small number of patients who fail to respond adequately to a gluten-free diet and they require therapy with glucocorticoids or immunosuppressive drugs. In visitors to the tropics, the onset of severe diarrhoea may be sudden and accompanied by fever. When the disorder becomes chronic, the features of megaloblastic anaemia (vitamin B12 and folic acid malabsorption) and other deficiencies, including ankle oedema, glossitis and stomatitis, are common. The differential diagnosis in the indigenous tropical population is an infective cause of diarrhoea. These arise because of deconjugation of bile acids, which impairs micelle formation, and because of bacterial utilisation of vitamin B12. Disorders that impair the normal physiological mechanisms controlling bacterial proliferation in the intestine predispose to bacterial overgrowth (Box 21. The most important are loss of gastric acidity, impaired intestinal motility and structural abnormalities that allow colonic bacteria to gain access to the small intestine or provide a secluded haven from the peristaltic stream. In systemic sclerosis, bacterial overgrowth arises because the circular and longitudinal layers of the intestinal muscle are fibrosed and motility is abnormal. Metabolic bone disease, folate or iron deficiency, coagulopathy and small bowel lymphoma are more common. A course of broad-spectrum antibiotic for 2 weeks is the first-line treatment, although there is no consensus on agent or dose. Examples include tetracycline (250 mg 4 times daily), metronidazole (400 mg 3 times daily), amoxicillin (250 mg 3 times daily) or ciprofloxacin (250 mg twice daily). If breath testing reveals high methane production, addition of neomycin (500 mg twice daily) may be beneficial. Some patients require up to 4 weeks of treatment and, in a few, continuous rotating courses of antibiotics are necessary. Intramuscular vitamin B12 supplementation may be needed in chronic cases, as the bacteria utilise vitamin B12. In most patients, pharmacological doses of folic acid (5 mg daily) improve symptoms and jejunal morphology. In some cases, treatment must be prolonged before improvement occurs and occasionally patients must leave the tropics. Jejunal contents for bacteriological examination can also be aspirated at endoscopy but laboratory analysis requires anaerobic and aerobic culture techniques. Bacterial overgrowth can also be diagnosed non-invasively using hydrogen breath tests, although they lack sensitivity.
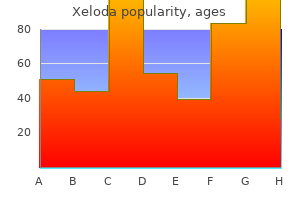
Cheap 500 mg xeloda with mastercard
Statement of the American Society of Human Genetics on cystic fibrosis carrier screening womens health october 2014 order xeloda 500mg mastercard. Preconception cystic fibrosis carrier couple screening: impact, understanding and satisfaction. X-linked (recessive) hypomaturation amelogenesis imperfecta: a prosthodontic, genetic and histopathologic report. Choroideremia: a clinical and genetic study of 84 Finnish patients and 126 female carriers. Lupus erythematosus-like oral mucosal and skin lesions in a carrier of chronic granulomatous disease. Clinical features of female heterozygotes in the X-linked mixed deafness syndrome (with perilymphatic gusher during stapes surgery). Development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies. Muscle X-inactivation patterns and dystrophin expression in Duchenne muscular dystrophy carriers. X-chromosome methylation in manifesting and healthy carriers of dystrophinopathies: concordance of activation ratios among first degree female relatives and skewed inactivation as cause of the affected phenotypes. Evidence for preferential X-chromosome inactivation in a family with Fabry disease. Congestive heart failure with rhabdomyolysis and acute renal failure in a manifesting female carrier of Duchenne muscular dystrophy with duplication of dystrophin gene. Neonatal hyperbilirubinaemia in heterozygous glucose-6-phosphate dehydrogenase deficient females. Ocular phenotypes associated with two mutations (R121W, C126X) in the Norrie disease gene. Fatal clinical course of ornithine transcarbamylase deficiency in an adult heterozygous female patient. Prenatal counseling in heterozygotes for ornithine transcarbamylase deficiency in an adult heterozygous female patient. Contribution to carrier detection and genetic counseling in X linked retinoschisis. Pyridoxine responsive hereditary sideroblastic erythropoiesis and iron overload: two microcytic subpopulations in the affected male, one normocytic and one microcytic subpopulation in the obligate female carrier. Familial occurrence of severe ulnar aplasia and lobster claw feet: a new syndrome. Comparison of X-chromosome inactivation in Duchenne muscle/myocardium-manifesting carriers, nonmanifesting carriers and related daughters. Adherence to American Academy of Pediatrics recommendations for cardiac care among female carriers of Duchenne and Becker muscular dystrophy. Reproductive choices and obstetrical experience in Dutch carriers of haemophilia A and B. Prenatal diagnosis for haemophilia: a nationwide survey among female carriers in the Netherlands. Pregnancy complications and obstetric care in women with inherited bleeding disorders. The risk of recurrent venous thromboembolism in patients with an Arg5066Gln mutation in the gene for factor V (factor V Leiden). Autosomal dominant polycystic kidney disease: new information for genetic counseling. Mutations in the cationic trypsinogen gene are associated with recurrent acute and chronic pancreatitis. Race, consanguinity and social features in Birmingham babies: a basis for prospective study. Consanguinity, fertility, reproductive wastage, infant mortality and congenital malformations in Jordan. Frequency and pattern of distribution of congenital anomalies in the newborn and associated maternal risk factors. Pathogenic variants for Mendelian and complex traits in exomes of 6,517 European and African Americans: implications for the return of incidental results. Quality in genetic counseling for presymptomatic testing-clinical guidelines for practice across the range of genetic conditions. A worldwide assessment of the frequency of suicide, suicide attempts or psychiatric hospitalization after predictive testing for Huntington disease. Predictive testing for Huntington disease: are we ready for widespread community implementation Protocol for genetic testing in Huntington disease: three years of experience in Minnesota. Attitudes of at-risk and affected individuals regarding presymptomatic testing for autosomal dominant polycystic kidney disease. Presymptomatic testing for adult onset polycystic kidney disease in at-risk kidney transplant donors. Attitudes towards cancer predictive testing and transmission of information to the family. Does the karyotype of a spontaneous abortion predict the karyotype of a subsequent abortion Paradoxical effects of repeat interruptions on spinocerebellar ataxia type 10 expansions and repeat instability. Corticobasal and ataxia syndromes widen the spectrum of C9orf72 hexanucleotide expansion disease. Databases of genomic variation and phenotypes: existing resources and future needs. The relationship between genotype and phenotype in cystic fibrosis: analysis of the most common mutation. Cystic fibrosis with three mutations in the cystic fibrosis transmembrane regulator gene. Parental somatic mosaicism is under recognized and influences recurrence risk of genomic disorders. Alternative explanations for recurrent achondroplasia in siblings with normal parents. Association of pigmentary anomalies with chromosomal and genetic mosaicism and chimerism. Genetic counseling on brittle grounds: recurring osteogenesis imperfecta due to parental mosaicism for a dominant mutation. Molecular and clinical correlations in spinocerebellar ataxia 2: a study of 32 families. A history of miscarriages and mild prognathism as possible mode of presentation of mosaic trisomy 18 in women. Variables influencing pregnancy termination following prenatal diagnosis of fetal chromosome abnormalities. The impact of personal loss on the experience of health professions: graduate students in end-of life and bereavement care. Disability training in the genetic counseling curricula: bridging the gap between genetic counsellors and the disability community. The psychosocial sequelae of a second-trimester termination of pregnancy for fetal abnormality. The psychic costs of empathic engagement: personal and demographic predictors of genetic counselor compassion fatigue. Developmental tasks of childhood and adolescence: implications for genetic testing. Points to consider: ethical, legal and psychosocial implications of genetic testing in children and adolescents. Fetal and perinatal autopsy in prenatally diagnosed fetal abnormalities with normal karyotype. Parental grief and memento mori photography: narrative, meaning, culture and context. Support after perinatal death: a study of support and counseling after bereavement. Preparing individuals to communicate genetic test results to their relatives: report of a randomized control trial.
Indian Head (Echinacea). Xeloda.
- What is Echinacea?
- Are there safety concerns?
- Is Echinacea effective?
- Urinary tract infections (UTIs), migraine headaches, chronic fatigue syndrome (CFS), eczema, hayfever, allergies, bee stings, attention deficit-hyperactivity disorder (ADHD), influenza (flu), and other conditions.
- What other names is Echinacea known by?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96942
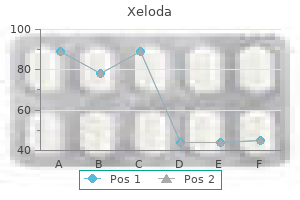
Order xeloda line
A particularly useful database for evaluating the prevalence of second-trimester chromosomal abnormality is a European collaborative study involving 52 women's health clinic toronto abortion purchase 500mg xeloda otc,965 amniocenteses performed on women aged 35 or more (Table 4. Based on amniocentesis data, it would appear that the frequency of structural chromosomal abnormalities is independent of maternal age. The frequency of Robertsonian translocations appeared to be underestimated, probably because of underreporting. These data show maternal age-specific prevalence for chromosomal abnormalities higher than that seen at amniocentesis (Table 4. Data from spontaneous abortuses Major chromosomal abnormalities have been found in nearly one half of all first-trimester spontaneously aborted fetuses. Of these, 55 percent were autosomal trisomies, 16 percent were 45,X, 20 percent were polyploidies, and 8 percent had other anomalies, such as a structural aberration, mosaicism, double trisomies, monosomy 21 or other complex karyotypes. Among the autosomal trisomies, any chromosome can be involved but trisomy 16 accounts for 25 percent of the cases (Table 4. The frequency of autosomal trisomies in spontaneous abortions increases with maternal age. Chromosomal abnormalities were found in 5 percent of the 3,237 specimens that included both complete and incomplete tissues and in 1. It is likely that the incomplete specimens contained a significant number of "blighted ova," either with no embryo or with a stunted embryo. Stillbirth is defined as the birth of a dead fetus during the late second or the third trimester of pregnancy (gestational age > 20 weeks whereas neonatal death refers to death occurring within Table 4. To provide adequate counseling for parents, all cases of stillbirth and neonatal death must be properly investigated. Thus, cytogenetic evaluation is an important component of perinatal autopsy (see Chapter 1). These frequencies of chromosomal abnormality in stillbirths and neonatal deaths are approximately 10 times higher than those in newborns. Individual patients may have multiple indications for prenatal cytogenetic diagnosis. However, it should be noted that some trisomy 21 and trisomy 13 cases will be due to the unbalanced segregation of a Robertsonian translocation and therefore karyotyping may be preferred because in most cases it will be the most efficient way to establish recurrence risk. It has been shown that there is a higher frequency of aneuploidies and dygynic triploidy in cases with low fetal fraction. These same markers can also be used to screen for trisomy 18 and the algorithm can be extended to trisomy 13. Different screening programs choose different criteria to identify their high-risk (screen-positive) groups and results may be quoted on the basis of first-trimester, second-trimester or term risk. For cases in which an anomaly is identified by ultrasound, amniocentesis should be considered. Abnormal ultrasound findings A large number of studies evaluated the risk for a chromosomal abnormality associated with the ultrasound identification of a fetal anomaly (see Chapter 13). They do, however, provide some indication of the magnitude of risk together with a guide to the most common chromosomal abnormalities seen. Much more data are needed to firmly establish that some of the rarer of the copy number variants identified are indeed the cause of the specific fetal abnormalities seen by ultrasound. Cystic hygromas are fluid accumulations in the lymphatics and are frequently associated with excess fluid in other tissues (nonimmune hydrops). Among second-trimester fetuses with cystic hygromas, only 37 percent show a normal karyotype. Noonan syndrome may also account for a relatively high proportion of cystic hygromas. A distinction has been drawn between cystic hygroma (bilateral, septated, cystic structures) and nuchal edema (subcutaneous fluid accumulation). Aneupl/ Total (%) 90/373 (24) 8/21 (38) Overall frequencya Notes: a Combined isolated and/or multiple ultrasound abnormalities. The types of cardiac defects found with 22q11 deletion may not be limited to conotruncal defects, and Manji et al. A meta-analysis indicated that approximately 4 percent of cases with a cardiac defect will have a 22q11. Intrauterine growth restriction, in the absence of any other biochemical or screening tests, will occasionally signal the presence of pregnancies affected by trisomies 13 and 18, but the finding is not a strong indicator for trisomy 21. Comparable levels of risk are associated with an ultrasound finding of microcephaly. Other anomalies that can be considered to be associated with a high risk for fetal aneuploidy include facial clefts, duodenal atresia ("double bubble" anomaly), some limb anomalies, and omphalocele (but not gastroschisis). There is also a high risk for aneuploidy when femur length, humerus length, or both, are shorter than that expected for the gestational age. Estimates for the risk for fetal aneuploidy when "echogenic bowel" is observed have been somewhat variable, probably reflecting the variable criteria used to define hyperechogenicity and ascertainment bias. Moderate risks for fetal aneuploidy can also be assigned when there is ultrasound detection of renal anomalies (including hydronephrosis), oligohydramnios or polyhydramnios, hydrocephaly/ ventriculomegaly, absent or hypoplastic nasal bone, aberrant right suclavian artery, and diaphragmatic hernia (in association with other anomalies). However, each of these findings has much greater significance when they are observed in the presence of additional ultrasound anomalies. The finding of any ultrasound anomaly, including these common low-risk factors, should prompt a thorough examination to determine whether additional anomalies are present. The "genetic sonogram" or "anomaly scan" In the second trimester, an ultrasound exam to evaluate the presence or absence of major structural abnormality or "soft" ultrasound markers is sometimes used to either increase or decrease the risk for trisomy 21 following a conventional screening test. Detection rate can be improved though the provision of the ultrasound exam to women who have both intermediate and high risks following the serum test. However, there are some situations where the genetic sonogram can still have some clinical utility. Moreover, the association between maternal age and fetal chromosomal abnormality can be a source of considerable anxiety, and a negative screening result is sometimes not sufficiently reassuring. There may be a minimal paternal age effect for trisomy 21 and Klinefelter syndrome but the relative risk appears much less than that associated with advanced paternal age and autosomal dominant diseases. Translocations with both 2:2 and 3:1 modes of segregation are considered together. Unbalanced forms all arise through 3: 1 segregation with +der(22) in these offspring. They are associated with intellectual disability but do not constitute a group of well defined syndromes. Genetic counseling for couples where there is advanced paternal age, typically 40 years or more at the time of conception, has been recommended. Carrier of a balanced structural rearrangement Although this indication represents only a small proportion (usually less than 5 percent) of prenatal diagnosis patients, the risk that these parents will bear offspring with an unbalanced chromosome complement can be high. The risk depends on (i) which parent is the carrier, (ii) the type of rearrangement, (iii) the method of ascertainment, (iv) the chromosome involved, and (v) the specific chromosome breakpoints. Reciprocal translocation Reciprocal translocation is the result of chromosome breaks in two autosomal chromosomes and the subsequent exchange of chromosome segments. In an early report of 609 prenatal diagnoses performed in cases in which one parent was a carrier of an apparently balanced reciprocal transloca- tion, 71 fetuses (11. Both the earlier data206, 208 and the updated data207 showed that the risk rates for unbalanced segregants diagnosed through amniocentesis are inversely related to the size of the chromosome imbalance. It is likely that many larger imbalances are associated with lethality and early embryonic or fetal loss. Individual translocation families are rarely large enough for reliable estimates of risks but data have been collected for different families with somewhat similar breakpoints. The number of observations in any particular breakpoint region may be small, so the confidence interval for the risk estimate may be very wide. Assumptions may also need to be made when there are two chromosome segments that are involved in the imbalance. Given these uncertainties, the risks associated with imbalance as presented in Table 4. These risks would not be applicable to a subtle subtelomeric translocation where there might be a milder phenotype and little selection against the potential chromosome imbalance. Male carriers of these translocations were far fewer than female carriers, suggesting an effect on male fertility.
Order generic xeloda on line
Parental decisions following the prenatal diagnosis of sex chromosome abnormalities molar pregnancy buy generic xeloda 500 mg. Parental decisions following prenatal diagnosis of chromosomal abnormalities: implications for genetic counseling practice in Japan. Factors influo a encing parental decision making in prenatal diagnosis of sex chromosome aneuploidy. Probes were initially labeled with biotin or digoxigenin and detected by fluorochrome-labeled antibodies; however, today, they are labeled directly with a fluorochrome. The use of fluorescent microscopy allows the detection of more than one probe, each labeled with a different color. The technology has now advanced so that combinatorial fluorescence with 24 different colors can be visualized on the same metaphase spread, thereby highlighting each chromosome pair. Interphase cells can be derived from cultured or direct (noncultivated) cells; preparations from tissue specimens may be examined either as dispersed cells or still in the original tissue architecture. The advent of molecular cytogenetics since the early 2000s has revolutionized both the research and clinical studies of chromosomes. The applications include studies of both metaphase chromosomes and interphase cells. Fluorescence in situ hybridization Genetic Disorders and the Fetus: Diagnosis, Prevention, and Treatment, Seventh Edition. The prototypic example of a contiguous gene syndrome was described by Francke et al. Several syndromes are associated with a characteristic deletion of chromosome 22 (22q11. Williams syndrome is a developmental disorder involving the central nervous system and vascular connective tissue. In these populations the clinical phenotype dictates which probes should be tested, and if patients should be referred to rule out a syndrome because of specific features. All the probes used postnatally can be used prenatally; however, lack of prenatal ascertainment of these syndromes precludes their use in most prenatal cases. One chromosome 17 shows two hybridization signals, one with the probe to the critical region and one with a control probe; therefore, this chromosome is normal. However, the other chromosome 17, denoted by an arrow, hybridizes only the control probe. One microdeletion that has been most frequently and successfully prenatally detected is the deletion of chromosome 22q11. This deletion is most often detected prenatally because of the presence of a conotruncal heart defect (tetralogy of Fallot, interrupted aortic arch); it has also been seen in association with uropathy and polyhydramnios, as well as being studied because of the presence of a familial deletion. However, they also 318 Genetic Disorders and the Fetus looked for a number of additional features in these fetuses. All nine cases were ascertained because of a low or absent maternal serum unconjugated estriol (uE3), which has been associated with placental sulfatase deficiency. However, as more microdeletions and microduplications have been delineated, especially with the use of array analysis, and their prenatal detection understood, the microdeletion methodology has been used more frequently. This is especially true for prenatal diagnostic studies in which the specimens cannot be analyzed easily with high-resolution procedures. However, even high-resolution analysis is not always sufficient for the interpretation of small structural rearrangements or complex karyotypes. A variety of studies since the mid-2000s have shown the importance of cryptic subtelomeric rearrangements in postnatal studies. These telomeric regions are thought to be gene rich and the loss of these regions is correlated with dysmorphic features and mental retardation. In 1996 two groups generated a complete set of unique sequence telomeric probes for the submicroscopic detection of subtelomeric chromosomal abnormalities. A probe for each chromosome arm has been developed, with a few exceptions; there are no probes for the individual acrocentric short arms, except for chromosome 15, and no unique probes for Xp and Yp because they share similar sequences, as do Xq and Yq. Although there is more than one technology, the most common is to use probes which use a total of 15 probe hybridizations per study. Though the postnatal studies are time consuming, this would create considerably more work on prenatal studies. Although the majority of subtle/cryptic rearrangements do involve the telomeric regions, there have also been a number of cryptic abnormalities involving interstitial regions. However, cryptic deletions associated with specific chromosomal rearrangements can possibly be delineated prenatally. Additional work, involving multiple cell harvests and additional chromosome banding techniques, together with a high degree of analytical skill at the microscope, was necessary for interpretation of these subtle rearrangements. These obstacles were especially formidable in the prenatal diagnostic arena, in which time is of the essence. This is especially true when a specific abnormality is detected prenatally and quick clarification is needed. The technology continues to improve and more probes have become available as the genome has been sequenced; therefore the delineation of cryptic aberrations has continued to expand. Although this translocation could be detected with standard banding, its elucidation in a prenatal specimen would have been problematic. Prenatal cytogenetic analysis of a subsequent pregnancy revealed what appeared to be a der(16) and an unbalanced karyotype. This impression was easily confirmed using chromosomes 15 and 16 specific painting probes. Fluorescence in situ hybridization analysis allowed for much more expeditious handling of this prenatal diagnostic case and in most cases similar rearrangements could be even more easily delineated using subtelomeric probes. Identification of marker chromosomes Determining the origin of chromosomal material that cannot be identified by conventional banding. Classification of such marker chromosomes is important for phenotype/karyotype correlations, which is imperative for proper counseling (see Chapter 4). Additional samples are not needed because unstained slides or fixed pellets are usually available and amenable to analysis. If necessary, previously banded slides can be used immediately, saving time in prenatal cases. The autosomal markers can be further subdivided into satellited or nonsatellited markers. The metaphase spread is initially analyzed with G banding to determine whether the marker is a satellited or nonsatellited derivative of an autosomal chromosome or if it is derived from a sex chromosome. When possible, a multiple-color approach for this analysis is taken, using two or three colors. This approach allows for the conservation of slides and material and, more importantly, permits a more immediate answer. Fourteen of the 16 pregnancies with Y-derived chromosome markers proceeded to term, with 13 resulting in the birth of a male infant. Twelve of these males appeared normal at birth; one had hypospadias and seminiferous tubules without germinal cells. Less follow up was available for the X-derived markers; however, one of these pregnancies ended as a stillbirth and several appeared normal at birth or showed phenotypic features associated with Turner syndrome. Approximately 17 percent of satellited markers are derived from chromosome 13 or 21, 24 percent from chromo- some 14 or 22, and 59 percent from chromosome 15. The presumption is that a monocentromeric, bisatellited marker consists of only repetitive sequences and has no phenotypic effects. However, dicentric markers with euchromatic material between the two centromeres may have a deleterious phenotypic effect. If a marker is dicentric, or monocentric and monosatellited, we then evaluate it with single-copy specific probes.
Purchase xeloda with american express
Rarely menopause essential oils buy cheapest xeloda and xeloda, it presents with sudden painless visual loss in the absence of raised inflammatory markers. Diagnosis can be made by demonstrating choroidal shutdown on fluorescein angiography. Some symptoms associated with visual loss require urgent ophthalmological assessment (Box 27. The presence of watering or watery discharge is not a discriminatory feature, and over-reliance on this symptom often results in anterior uveitis being misdiagnosed as viral conjunctivitis. Blurred vision describes the situation in which patients are able to see what they are looking at, but what they are looking at is out of focus. The most common cause of intermittent blurred vision is dry eye; the most common cause of permanent blurred vision is cataract. If blurred vision is worse in the morning and eases as the day progresses, this suggests macular oedema. Pain/headache Blurred vision ks A flickering light sensation is indicative of photoreceptor activity, either through traction, as in the setting of posterior vitreous detachment, or inflammation, as in the setting of autoimmune or paraneoplastic retinopathy. Rarely, photopsia is a symptom of occipital lobe epilepsy, in which case there is usually an accompanying homonymous hemianopia. A significant proportion of people are allergic to topical chloramphenicol, a first-line treatment for many minor ocular ailments. Patients with dry eye may complain of a foreign body or gritty sensation in the eye or intermittent visual blurring, triggered by reduced blinking, as occurs when reading or when concentrating on a distant object, such as the television. Photophobia may also be a feature of meningitis, usually with accompanying neck stiffness and headache (meningism, p. Glare is a common early feature of cataract, particularly triggered by oncoming car headlights when driving at night. It may also be an issue where there is insufficient melanin in the retinal pigment epithelium. If surgery is not an option, or while surgery is awaited, the symptom of glare may be reduced by wearing a broad-brimmed hat. Less commonly, it can be caused by epiretinal membrane formation, where posterior hyaloid surface scarring causes foveal traction. Usually with distortion, objects are not only misshapen but also smaller (micropsia), due to the photoreceptors being pulled apart. Compensatory increased innervation to the superior rectus and the levator palpebrae superioris, as well as direct inflammation, leads to eyelid retraction. In thyrotoxicosis, increased sympathetic nervous activity leads to bilateral eyelid retraction. The cardinal features are inflammation of the lacrimal gland, its conjunctival accessory glands and the parotid gland, leading to hyposecretion of tears and saliva. Involvement of the lacrimal gland alone causes keratoconjunctivitis sicca, a syndrome of dry eyes and corneal and conjunctival irritation. Keratoconjunctivitis sicca, however, can also be caused by reduced function of the lacrimal glands and/or lacrimal ducts from other causes. If these measures are insufficient, tear drainage may be reduced with surgical options such as punctal plugs and punctal occlusion. In structures in direct contact with the environment, particularly the cornea and the conjunctiva, inflammation is most likely to be caused by infection. In other structures, such as the uveal tract and sclera, inflammation is more likely to be caused by autoimmune conditions, although it may also be a manifestation of infection or malignancy. Although the latter conditions may present with indicative ocular signs, their presence is often appreciated only retrospectively, after failure to respond to immunosuppression. Most non-infective forms of ocular inflammation are idiopathic; all are more common in the presence of other autoimmune conditions. Some may be directly associated but asynchronous in disease activity, such as the anterior uveitis of ankylosing spondylitis (p. The most common cause of transient visual loss is the aura of migraine, usually a positive phenomenon with the object of regard seemingly hidden by something in the way, rather than a negative phenomenon in which part or all of what is being looked at is missing. With positive visual phenomena the obstruction is often white or coloured, expanding across the visual field, or in a constant position but shimmering. Negative visual phenomena are a cardinal feature of ocular, usually retinal, ischaemia, with complete absence of vision (blackness) occupying part or all the visual field. Transient ocular ischaemia is usually embolic in nature but is occasionally seen in giant cell arteritis, where it suggests critical optic nerve ischaemia. Permanent monocular negative visual phenomena usually indicate previous optic nerve or retinal infarction. Tiny negative visual phenomena may also be seen in capillary disorders such as diabetic retinopathy, where patchy macular capillary occlusion may, for instance, cause letters to be missing from words on reading. Proptosis is a sign of retro-orbital expansion and may be intraconal or extraconal. When expansion is within the cone of extraocular muscles, then movement forwards will be in line with the visual axis. The primary clinical concern is whether vision is at risk due to optic nerve compression or corneal exposure. Instead, restricted ocular movements make patients move their head en bloc when looking at objects deviating from the primary position of gaze. To the patient, however, the overarching concern is often the change in appearance. It may be classified according to speed of onset, location, specific features, or aetiology (Box 27. Active tuberculosis may present with an occlusive vasculitis or serpiginous (snake-like) choroiditis emanating from the optic disc. Often confused with scleritis, although usually less symptomatic, the diagnostic topical application of phenylephrine turns the inflamed episclera white but has no effect on the redness of scleritis. Diagnosis of anterior scleritis is usually straightforward, with the eye showing diffuse or nodular erythema (although it may have to be searched for under the eyelids). Posterior uveitis is often accompanied by reduced vision and oedema of the retina, choroid and extraocular muscles. White patches of necrosis (pallor) within the erythema are an ominous sign, indicative of systemic vasculitis. Non-necrotising scleritis is commonly idiopathic but may be associated with other autoimmune conditions, particularly rheumatoid arthritis and inflammatory bowel disease. Some patients with recurrent episodes of scleritis, or in whom inflammation is gradual and prolonged, may develop scleral thinning (scleromalacia), revealing the underlying blue choroid. Posterior complications can also develop, predominantly macular oedema, the main cause of visual impairment in all forms of uveitis. With intermediate uveitis, inflammation occurs at the pars plana, with most symptoms, predominantly floaters, being a result of inflammation of the vitreous base. Unlike anterior uveitis, pure intermediate uveitis is not associated with iris inflammation; instead, white blood cells are seen predominantly in the anterior vitreous, with a lesser amount overspilling into the anterior chamber. Topical therapy is ineffective, as it does not penetrate beyond the anterior chamber, but symptoms of floaters are not often sufficient to justify systemic immunosuppression. In some cases, vitritis (vitreous inflammation), or more commonly macular oedema, may cause visual impairment. It may be directly associated with inflammatory disorders in which immune complexes are formed, particularly rheumatoid arthritis, systemic lupus erythematosus and granulomatosis with polyangiitis. Systemic immunosuppression is always required but topical glucocorticoids should be used cautiously due to the risk of aggravating keratolysis (corneal thinning). Secondary infection should be prevented with topical antibiotics and attention should be paid to corneal hydration, through the use of artificial tears and lubricants. More common causes of peripheral corneal ulceration are blepharitis and acne rosacea, causing ocular irritation rather than frank pain. Hypersensitivity to staphylococcal exotoxin leads to stromal infiltrate adjacent to , but sparing, the limbus (marginal keratitis). Resolution of this self-limiting condition can be assisted by the use of topical chloramphenicol, with or without topical glucocorticoids. Prevention is through management of the underlying condition, usually with ocular lid hygiene for simple blepharitis and metronidazole gel for rosacea.
Order cheapest xeloda and xeloda
Regulation of growth and gene activity in euploid hybrids between human neonatal fibroblasts and epithelioid amniotic fluid cells best women's health tips generic xeloda 500mg mastercard. Cell type identification via one and two-dimensional electrophoresis of clonal whole cell homogenates. Cultured human amniotic fluid cells characterized with antibodies against intermediate filaments in indirect immunofluorescence microscopy. Characterization of cells of amniotic fluids by immunological identification of intermediate-sized filaments: presence of cells of different tissue origin. Studies on the origin of human amniotic fluid cells by immunofluorescent staining of keratin filaments. Epithelial character and morphologic diversity of cell cultures from human amniotic fluids examined by immunofluorescence microscopy and gel electrophoresis of cytoskeletal proteins. Intermediate filament cytoskeleton of amnion epithelium and cultured amnion epithelial cells: expression of epidermal cytokeratins in cells of a simple epithelium. Antenatal detection of neural tube defects: comparison of biochemical and immunofluorescence methods. Cultivated epithelial-like cells and fibroblasts from amniotic fluid: their relationship to enzymatic and cytologic analysis. Lysosomal enzyme activities in different types of amniotic fluid cells measured by microchemical methods, combined with interference microscopy. Biochemical and biological problems and pitfalls of cell culture for prenatal diagnosis. Growth of cells in hormonally defined media (Cold Spring Harbor Conferences on Cell Proliferation). Human epithelial cells cultured from urine: growth properties and keratin staining. Sensitivity of chromosomal mosaicism detection by different tissue culture methods. First trimester amniocentesis between the seventh and 13th weeks: evaluation of the earliest possible genetic diagnosis. A culture vessel for amniotic fluid cells allowing faster preparation of chromosome slides. Improvements in cytogenetic slide preparation: controlled chromosome spreading, chemical aging and gradual denaturing. Exclusion of chromosomal mosaicism in amniotic fluid cultures: efficacy of in situ versus flask techniques. Exclusion of chromosomal mosaicism in amniotic fluid cultures: determination of number of colonies needed for accurate analysis. Culturing and robotic harvesting of bone marrow, lymph nodes, peripheral blood, fibroblasts, and solid tumors with in situ techniques. Effect of pre-amniocentesis uterine manipulation on amniocyte concentration and culture duration: a randomized, clinical trial. Amniofiltration in the first trimester: feasibility, technical aspects 170 Genetic Disorders and the Fetus 640. Early sonographically guided amniocenteses with filtration technique: follow-up on 249 procedures. Early filtration amniocentesis for further investigation of mosaicism diagnosed by chorionic villus sampling. Amniotic fluid fibronectin in normal pregnancy and in pregnancies with anencephalic fetus. Stimulation of human amniotic fluid cell proliferation and colony formation by cell plating on a naturally produced extracellular matrix. Influence of extracellular matrix on the proliferation of human amniotic fluid cells in vitro. Decreased oxygen supply enhances growth in culture of human midtrimester amniotic fluid cells. The effect of oxygen tension on colony formation and cell proliferation of amniotic fluid cells in vitro. Effects of fibroblast growth factor and epidermal growth factor on the rate of growth of amniotic fluid-derived cells. Freezing and thawing serum and other biological materials: optimal procedures minimize damage and maximize shelf-life. Human amniotic fluid cells grown in a hormone-supplemented medium: suitability for prenatal diagnosis. The pipette method: a new rapid technique for chromosome analysis in prenatal diagnosis. Routine use of Chang medium for prenatal diagnosis: improved growth and increased chromosomal breakage. American College of Medical Genetics Standards and Guidelines for Clinical Genetics Laboratories. Variables influencing growth and morphology of colonies of cells from human amniotic fluid. Evaluation of a red blood cell lysis procedure for culture of cells from blood-contaminated amniotic fluid. Isolation of human multipotent mesenchymal stem cells from secondtrimester amniotic fluid using a novel two-stage culture protocol. Molecular and proteomic characterization of human mesenchymal stem cells derived from amniotic fluid: comparison to bone marrow mesenchymal stem cells. Amniotic fluid-derived mesenchymal stem cells: characteristics and therapeutic applications. Amniotic fluid as a novel source of mesenchymal stem cells for therapeutic transplantation. Xenografted human amniotic fluid-derived stem cell as a cell source in therapeutic angiogenesis. Amniotic fluid-derived stem cells for cardiovascular tissue engineering applications. Potential antitumor therapeutic strategies of human amniotic membrane and amniotic fluid-derived stem cells. Fetal mesenchymal stem cells: isolation, properties and potential use in perinatology and regenerative medicine. Amniotic fluid and placental membranes: unexpected sources of highly multipotent cells. Determination of amniotic fluid acetylcholinesterase activity in the antenatal diagnosis of foetal malformations: the first ten years. Alphafetoprotein detection of neural tube defects and the impact of standard ultrasound. Synthesis of alphafetoprotein by liver, yolk sac, and gastrointestinal tract of the human conceptus. Alphafetoprotein and acetylcholinesterase activity in firstand early second-trimester amniotic fluid. Amniotic fluid alpha-fetoprotein measurement in antenatal diagnosis of anencephaly and open spina bifida in early pregnancy. Radioimmunoassay of alphafetoprotein in hematoma, other liver diseases and pregnancy. Routine measurement of amniotic fluid alpha-fetoprotein and acetylcholinesterase: the need for a reevaluation. Amniotic fluid alpha-fetoprotein concentrations in twin gestations: dependence on placental membrane anatomy. Alpha fetoprotein and acetylcholinesterase levels in twins discordant for neural tube defects: dependence on type of fetal membranes. Biamnial elevated alpha-fetoprotein and positive acetylcholinesterase in twins, one with anencephaly. Acetylcholinesterase activity in amniotic fluid of normal and anencephalic fetus in diamniotic twin pregnancy.
500mg xeloda amex
However menstruation 10 year old purchase xeloda on line, caution is required, particularly on the hands, as over-vigorous cryotherapy can lead to scarring, nail dystrophy and even tendon rupture. Periungual and subungual warts can be problematic and nail cutting and subsequent electrodessication may help. Several other therapies have been used for recalcitrant warts, including topical formaldehyde, podophyllotoxin, trichloroacetic acid, cantharidin, topical or systemic retinoids, intralesional bleomycin or interferon injections, and contact sensitisation with, for example, diphencyprone. Dermatophyte infections (ringworm) are extremely common and usually caused by fungi of the Microsporum, Trichophyton and Epidermophyton species. The fungi can originate from soil (geophilic) or animals (zoophilic), or be confined to human skin (anthropophilic). Dermatophyte infections usually present with skin (tinea corporis), scalp (tinea capitis), groin (tinea cruris), foot (tinea pedis) and/or nail (onychomycosis) involvement. Topical antifungals such as terbinafine or miconazole may suffice, although systemic treatment (terbinafine, itraconazole or griseofulvin) may be required for stubborn or extensive disease and scalp or nail involvement. Indeed, prolonged courses of systemic treatment may be needed for nail involvement. The capillary loops are evident Orf Orf is a parapoxvirus skin infection and is an occupational risk for those who work with sheep and goats. Itchy, erythematous plaques develop in the groins and extend on to the thighs, with a raised active edge. It typically presents as an area of scalp inflammation and scaling, often with pustules and partial hair loss. It typically presents as an itchy rash between the toes, with peeling, fissuring and maceration. Involvement of one sole or palm (tinea manuum) with fine scaling is characteristic of T. Pityriasis versicolor is a persistent, superficial skin condition caused by various species of the commensal yeast Malassezia, most commonly Malassezia globosa, but sometimes M. It is found more frequently in warmer, humid climates, and is usually more severe and persistent in the immunocompromised. It is characterised by scaly, oval macules on the upper trunk, usually hypopigmented but occasionally hyperpigmented. Treatment with selenium sulphide or ketoconazole shampoos and topical or systemic azole antifungal agents is usually effective, although recurrence is common because these yeasts are skin commensals, and maintenance topical therapy may be required. Infections are usually not serious, unless the patient is immunocompromised, in which case deeper tissues can be involved (p. The organism has a predilection for warm, moist environments and typical presentations are napkin candidiasis in babies, genital and perineal candidiasis, intertrigo and oral candidiasis. The diagnosis can be confirmed by microscopy and culture of skin swabs, and treatment is with topical or systemic antifungals, such as azoles. Typically, lesions are erythematous, annular and scaly, with a well-defined edge and central clearing. The degree of inflammation is dependent on the organism involved and the host immune response. Microsporum canis (from dogs) and Trichophyton verrucosum (from cats) are common culprits. Ill-advised use of topical glucocorticoids can modify the clinical presentation and increase disease extension (tinea incognito). Onychomycosis usually presents with yellow/brown nail discoloration, crumbling, thickening and subungual hyperkeratosis. Usually, some nails are spared, there is asymmetry and toenails are more commonly involved. In addition to systemic antifungals, short courses of systemic or topical glucocorticoid are often used in kerion on the basis of reducing inflammation and possible hair loss. Kerion is a boggy, inflammatory area of tinea capitis, usually caused by zoophilic fungi such as cattle ringworm (T. B A re sf re sf sf re mite still in its egg, seen on light microscopy of scrapings over a burrow. The consequences (whether acne is objectively severe or not) can be devastating, leading to embarrassment, school avoidance, and life-long effects on ability to form friendships, attract partners, and acquire and keep employment. It is extremely common, generally starts during puberty and has been estimated to affect over 90% of adolescents. It is usually most severe in the late teenage years but can persist into the thirties and forties, particularly in females (Box 29. Management is as for head and body lice and whole-body treatment should be undertaken. Sexual and other close contacts should also be treated and patients should also be screened for sexually transmitted diseases. It spreads in households and environments where there is intimate personal contact. The diagnosis is made by identifying the scabietic burrow (definition: a linear or curvilinear papule, caused by a burrowing scabies mite; p. The clinical features include secondary eczematisation elsewhere on the body; the face and scalp are rarely affected, except in infants. Even after successful treatment, itch can continue and occasionally nodular lesions persist. Topical treatment of the affected individual and all asymptomatic family members/physical contacts is required to ensure eradication. Two applications 1 week apart of an aqueous solution of permethrin or malathion to the whole body, excluding the head, are usually successful. Itch, excoriation (definition: a linear ulcer or erosion resulting from scratching) and secondary infection occur. Dry-cleaning and high-temperature washing or insecticide treatment of clothes are required. The diagnosis is confirmed by identifying the living louse or nymph on the scalp or on a black sheet of paper after careful fine-toothed combing of wet hair following conditioner application. Treatment is recommended for the affected individual and any infected household/school contacts. Eradication in school populations is difficult because of poor adherence and treatment resistance. Vaseline should be applied to eyelashes/brows twice daily for at least a fortnight. It is rare, usually affecting adult males, and most commonly occurs on trunk and upper limbs. It may be associated with hidradenitis suppurativa (a chronic, inflammatory disorder of apocrine glands, predominantly affecting axillae and groins), scalp folliculitis and pilonidal sinus. If comedones predominate, then topical benzoyl peroxide or retinoids should be used. It is an irritant, which may contribute to the therapeutic response, but this can be minimised by adjusting treatment regimes. Secondary causes and suspected underlying endocrine disease or virilisation should be investigated (p. Severity of acne is associated with sebum excretion rate, which increases at puberty. Both androgens and progestogens increase sebum excretion and oestrogens reduce it, but most patients with acne have normal hormone profiles. There may be a positive family history and there is high concordance in monozygotic twins, indicating that genetic factors are important, but the candidate genes are poorly defined. It usually affects teenage girls, and underlying psychological problems are common. Most patients with acne do not have an underlying endocrine disorder but acne is a common feature of polycystic ovary syndrome (p.
