

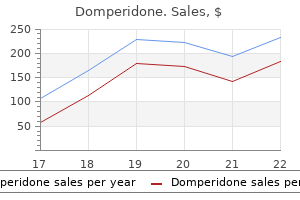
Order domperidone 10mg line
Skeletal maturity at onset of the adolescent growth spurt and at peak velocity for growth in height: a threshold effect? Application of the Gompertz curve to the observed pattern of growth in length of 48 individual boys and girls during the adolescent cycle of growth symptoms 3dpo buy generic domperidone line. Cross-sectional study of skeletal maturation in normal children from Nottingham and London. Interrelationships of skeletal maturation, sexual development and somatic growth in man. TannerΗhitehouse method of assessing skeletal maturity: problems and common errors. Skeletal maturity standards for French-Canadian children of school-age with a discussion of the reliability and validity of such measures. A comparison of skeletal growth and maturation in undernourished and well-nourished girls before and after menarche. Skeletal maturity and socio-economic status in Portuguese children and youths: the Madeira growth study. Skeletal development of Finnish children in the light of hand-wrist roentgenograms. Applicability of the Greulich and Pyle skeletal age standards to black and white children of today. Height, mass and skeletal maturity of elite Portuguese soccer players aged 11ͱ6 years. Skeletal age determinations in children of European and African descent: applicability of the Greulich and Pyle standards. Skeletal maturation of Hong Kong Chinese children in the first five years of life. Growth in some physical dimensions in relation to adolescent growth spurt among rural Indian children. Serum insulin-like growth factor-I, insulin-like growth factor binding protein-3, and the pubertal growth spurt in the female rhesus monkey. Volumetric thoracic growth in children with moderate and severe scoliosis compared to subjects without spinal deformity. Predicting scoliosis progression from skeletal maturity: reliability and validity of a simplified TannerWhitehouse classification system in girls with idiopathic scoliosis. Alman Genetic Aspects of Orthopaedic Conditions A genetic and molecular revolution is happening in medicine. Led by the Human Genome Project, genetic information and concepts are changing the way disease is defined, diagnosis is made, and treatment strategies are developed. The profound implications of actually understanding the molecular abnormalities of many clinical problems are affecting virtually all medical and surgical disciplines. Importantly, genetic technologies will increasingly drive biomedical research and the practice of medicine in the near future. It is important for those interested in the musculoskeletal system to be aware of the genetic cause of its inherited disorders in order to make appropriate referrals for genetic counseling and to refine the prognosis and natural history in each individual patient. Current management revolves around treatment to prevent or minimize medical complications, psychosocial support of patients and their families, and modification of the environment where appropriate. Gene discoveries will allow the development of tests to detect disease or to quantify the risk of disease. Furthermore, applying this knowledge is the best hope for developing strategies to modify the pathologic effect of the gene (drug therapy) or repair the gene (gene therapy) or for approaches to restore lost or affected tissue (tissue engineering). Instead of an empiric trial-and-error approach to therapy, it may become feasible to tailor treatment to the specific molecular malfunction. Given the large number of inherited musculoskeletal abnormalities and the power and speed of current genetic and developmental biology information, a few selected disorders are discussed in this chapter to illustrate specific concepts on the basic genetic concepts. It also discusses current classifications and clinical evaluation and provides a perspective about genetic counseling. Each species is different, and each reproduces itself faithfully: parent organisms hand down information specifying, in extraordinary detail, the characteristics that the offspring shall have. Hereditary phenomena have been of interest to humans long before biology or genetics existed as a scientific discipline. Ancient peoples improved crops and domesticated animals by selecting desirable individuals for breeding. However, the prevailing notion at the time was that the spermatozoon and the egg contained sampling of essences from the various parts of the parental bodies and at conception, they blended together. But there were many instances in which this mode of inheritance could not explain many of the observations on heredity. As a modern discipline, genetics began in the 1860s with the work of Gregor Mendel who performed a set of experiments that pointed to the existence of biological elements that we now call genes. The discovery of genes and the understanding of their molecular structure and function have been a source of profound insight into two of the biggest mysteries of biology: what makes a species what it is and what causes variation within a species? In addition, it has exploded our understanding of human diseases, specifically genetic disorders. Genetics took a major step forward with the notion that the genes, as characterized by Mendel, are part of specific cellular structures, the chromosomes. This fusion between genetics and cell biology is still an essential part of genetic analysis today and has tremendous implications in many fields including medical genetics (4Ͷ). Three fundamental properties are required of genes: (a) replication - hereditary molecules must be capable of being copied to ensure the continuation of a species from one generation to the next; (b) information storage - genes have the essential coding specifications to be translated to proteins and their by-products which constitute the building blocks of the organism; and (c) mutation - genes can change over time and this is the basis for variation within species and for evolution. The cells of most bacteria, algae, and fungi contain just one genome, that is, they are haploid. Interestingly, even closely related species with similar genome sizes can have very different numbers and sizes of chromosomes. Thus, there is no simple relationship between chromosome number, species complexity, and total genome size. Karyotype refers to the complement of chromosomes as visualized by cytogenetic analysis at mitosis. Human somatic cells contain two sets of 23 chromosomes, for a total of 46, referred to as euploidy. The maternal and paternal chromosomes of a pair are called homologous chromosomes (homologs). Each chromosome carries a different array of genes linearly arranged along the chromosome (each gene occupies a particular position or locus); therefore, genes are present twice (one allele coming from the mother and one from the father). When a cell divides, all chromosomes are replicated and each daughter cell contains the full complement of chromosomes. When a chromosome is replicated, all the genes in that chromosome are automatically copied along with it. In contrast, germ-line cells undergo meiosis during which the diploid number of 46 chromosomes is reduced to the haploid number of 23, including one of each of the autosomes and either the sex chromosome X or Y. The highly programmed chromosomal movements in meiosis cause the equal segregation of alleles into the gametes. For instance, during meiosis in a heterozygote A/a, the chromosome carrying A is pulled in the opposite direction from the chromosome carrying a, so half the resulting gametes carry A and the other half carry a. The random assortment of each of the chromosome pairs during meiosis is central to the inheritance pattern of single-gene disorders and some forms of chromosomal derangements. The force pulling the chromosomes to cell poles is generated by the nuclear spindle, a series of microtubules made of the protein tubulin. Microtubules attach to the centromeres of chromosomes by interacting with another specific set of proteins located in this area. The orchestration of these molecular interactions is complex, yet constitutes the basis of the laws of hereditary transmission in eukaryotes. A typical human somatic cell contains two of each of these chromosomes, plus two sex chromosomes - two X chromosomes in a female and one X and one Y in a male. This process occurs through an ordered series of stages, collectively known as cell cycle. Each of the four nucleotides is usually designated by the first letter of the base that it contains: A (adenine), G (guanine), C (cytosine), or T (thymine). The two nucleotide chains are held together by hydrogen bonds between the nucleotide bases. The biological role of most genes is to carry or encode information on the composition of proteins.
Cheap domperidone 10 mg
It is commonly associated with obesity keratin intensive treatment purchase domperidone 10mg mastercard, diabetes mellitus type 2, jejuno-ileal bypass surgery, parenteral nutrition, bacterial overgrowth of the small intestine, and various drugs. There is growing evidence that acquired mitochondrial electron transport abnormalities may underlie the oxidant stress generated and contribute to the intracellular accumulation of microvesicular fat. Circulating lipopolysaccharide and tumor necrosis factor- levels are elevated in several of the associated conditions and may contribute to both the oxidant stress and mitochondrial dysfunction. Use of antioxidant therapy for non-alcoholic steatohepatitis, including vitamin E, is discussed in Chapter 36. In one large series, 36% presented before 1 month of life, 44% between 1 month and 2 years, and 20% after 2 years of age. Within several years, whole genome or whole exome sequencing will be commercially available and will cost less than single gene sequencing costs today. Interpretation of genotyping results will be critical to making the proper diagnosis. A tiered approach to diagnosing hepatic mitochondrial disorders is recommended, as below, with initial screening tests (tier one), followed by either biochemical assays in affected tissues or genotyping for relatively common causative genes (tier two), followed by further molecular and biochemical evaluations (tier 3). Screening tests for respiratory chain defects Laboratory findings which suggest the presence of a respiratory chain defect are listed in Table 35. It should be stressed, however, that lactic acid and these ratios are not elevated in all patients with respiratory chain defects. After feeding or intravenous dextrose, the exaggerated paradoxical production of ketones is even more evident, as ketone production should normally decrease after meals (or glucose infusion) through the suppressive effect of insulin on ketogenesis. Therefore, to fully evaluate the patient, some have recommended that the concentration of these substrates and their molar ratios, as well as Diagnosis of respiratory chain disorders General clinical features and diagnostic approach Diagnosing a mitochondrial respiratory chain defect in patients with liver disease requires a high index of suspicion. Clinical findings that should suggest these disorders include (1) association of neuromuscular symptoms with liver dysfunction; (2) multisystem involvement in a patient with acute or chronic liver disease; (3) a rapidly progressive course of liver disease, particularly in the presence of lactic acidosis, hepatic steatosis, or ketonemia; and (d) onset of liver disease in the first 2 years of life. A more complete list of presenting clinical symptoms at various ages has been published by Munnich et al. Occasionally, it is necessary to load a fasted patient with oral glucose (2 g/kg) in order to provoke lactic acidemia and abnormal ratios if the values are normal under baseline conditions. Substrates and ratios should be measured every 15 minutes for 90 minutes after the load. Pitfalls in interpretation of these ratios include false positives in patients with systemic hypotension or with impaired ventilation. In addition, pyruvate and acetoacetate are less stable than lactate and -hydroxybutyrate, and artefacts of sample preparation or delayed processing may result in spuriously increased ratios. It should be pointed out that patients with the later presentation of liver dysfunction. AlpersΈuttenlocher syndrome) frequently do not have elevations of plasma lactate or increased plasma lactate to pyruvate and arterial ketone body ratios. Patients with pyruvate dehydrogenase deficiency generally have low plasma lactate to pyruvate ratios (<10). Renal tubular dysfunction can cause lower plasma lactate levels, thus making the lactate to pyruvate ratio less accurate. Proximal renal tubular dysfunction may lower plasma lactate and increase urinary lactate. It should be emphasized that these are screening tests and may not be abnormal if the respiratory chain defect is confined to one or two organs. Therefore, searching for dysfunction or abnormal histology/biochemistry of the target organs is also important. Definitive diagnostic tests Measurement of mitochondrial respiration If freshly obtained affected tissue and the laboratory expertise are available, analysis of oxygen consumption in mitochondrial-enriched fractions of tissues (liver, muscle, lymphocytes, fibroblasts) can be performed by polarographic studies in the presence of a series of respiratory substrates to define the site of the respiratory impairment [2,63]. Polarographic studies have the advantage of requiring relatively small amounts of tissue (100Ͳ00 mg muscle; 10 mL blood for circulating lymphocytes); however, these assays require fresh tissue for immediate isolation and analysis of mitochondria. Polarographic testing is available on fresh liver biopsy specimens at very few centers around the world and is impractical for evaluation of most patients. Histopathological investigations It is possible to detect deficiencies of subunits of several of the respiratory chain complexes by histochemical examination of biopsy material. Tissue specimens must be frozen immediately in liquid nitrogen-cooled isopentane for these studies. Histology and electron microscopy of muscle and liver may also reveal characteristic findings of respiratory chain disorders. Magnetic resonance imaging Magnetic resonance imaging may be useful in diagnosis of mitochondrial disease and in documenting whether the brain is involved if liver transplantation is being considered [66]. Cerebral lactate measurements, the end-product of non-oxidative metabolism (glycolysis), can be done using 1H magnetic resonance spectroscopy. Other features of energy metabolism, as measured with phosphorus-31 spectroscopy, may also have utility in characterizing mitochondrial disease. Enzymatic activity of respiratory chain complexes Direct measurement of the enzymatic activity of the mitochondrial respiratory chain complexes on frozen tissue is more commonly used to establish deficiency of the respiratory chain [64]. These studies can be performed on frozen samples of small biopsies of liver, muscle, kidney, myocardium, or other tissue since they do not require the isolation of mitochondrial fractions, and can be carried out on tissue homogenates, lymphocytes, or cultured skin fibroblasts. Tissue biopsy samples must be frozen immediately at the bedside or in the operating room and stored at 80у until analyzed. Measurement of respiratory chain enzyme activities separately or in groups is performed in specialized laboratories spectrophotometrically using specific electron acceptors and donors. In general, the tissues chosen for study should be those which clinically express the disease. It may also be useful to obtain skin fibroblasts and lymphocytes for testing, although a defect may be absent in these cells and confined to the involved organ. However, in most centers, these analyses usually require surgical liver or muscle biopsies. Normal values may have a wide range, so overlap may be found in affected patients. For this reason, some experts recommend expressing results as ratios of one protein complex activity to another to detect deficiencies of one enzyme that may be in the low to normal range [65]. Finally, blue native polyacrilamide gel electrophoresis of tissue can detect incompletely assembled respiratory chain subunits and may be helpful in establishing the diagnosis. Using these molecular techniques, specific mutations or deletions can be sought and even the entire mitochondrial genome can be sequenced. As stated, large genotyping panels of over 100 nuclear genes coding for mitochondrial proteins (using next-generation sequencing platforms) are now commercially available in some countries at great cost savings compared with genotyping individual genes and might be considered as a cost-effective way to evaluate patients. Acute metabolic acidosis is treated with the slow infusion of sodium bicarbonate intravenously, anemia or thrombocytopenia is treated with transfusions, and chronic pancreatic insufficiency may require the provision of exogenous pancreatic enzymes with meals. Acute liver failure is treated with balanced intravenous administration of dextrose and lipids, correction of acidosis, treatment of hyperammonemia, and attention to infectious complications. Chronic liver disease management may include the use of infant formula high in medium-chain triglycerides, a diet with at least 30ʹ0% fat, cornstarch to prevent hypoglycemia, evaluation and supplementation with fat-soluble vitamins (if cholestatic), routine immunizations (including hepatitis A and B vaccines), and monitoring for and treating complications of portal hypertension. Other mitochondrial strategies include supplementation with mitochondrial cofactors, scavengers of oxygen free radicals, and mitochondrial energy substrates, as well as avoidance of drugs and conditions known to have a detrimental effect on the respiratory chain [2]. Valproic acid should likewise be avoided if possible in these patients because of its effects on respiration and fatty acid metabolism. Pharmacologic support A variety of antioxidant compounds have been proposed as scavengers of electrons or oxygen free radicals, as promoters of electron transport, or as stimulators of mitochondrial respiration [69,70]. Using an animal model of liver injury that employed endotoxin-induced intrahepatic lipid peroxidation, exogenously administered ubiquinone prevented the marked reduction in hepatic levels of endogenous coenzyme Q, -tocopherol, and glutathione; suppressed lipid peroxidation; and increased the survival rate in endotoxemic mice. Similar effects of ubiquinone were seen in an animal model of liver ischemiaβeperfusion injury. Coenzyme Q analogues have also been shown to promote respiration in isolated hepatic and brain mitochondria and hepatic mitochondrial coenzyme Q levels have been shown to decrease after several weeks of bile duct ligation in the rat. However, supplementation has not Treatment of respiratory chain disorders Unfortunately, there is no ideal effective therapy for most patients with respiratory chain disorders, including those with liver failure and more slowly progressive liver disease. It is not clear that any currently available medical therapy significantly alters the course of severe disease; however, some patients have experienced improvement of neuromuscular symptoms with specific therapies. A recent Cochrane review of treatment strategies for mitochondrial disorders identified 12 studies that fulfilled the entry criteria [68]. The comparability of the reviewed studies was extremely low because of differences in the specific diseases studied, differences in the therapeutic agents used, dosage, study design, and outcomes. There was no clear evidence supporting the use of any intervention in mitochondrial disorders. A recommendation was made to test novel agents in homogeneous study populations with clinically defined primary end-points. In mitochondrial myopathies or cardiomyopathies, occasional patients have shown dramatic improvement in muscle strength and cardiac function after coenzyme Q supplementation.
Cheap domperidone online mastercard
After the index case in a maternal sibship there is an approximately 90% probability that each subsequent baby born to that mother will be affected [36] medicine 2355 buy genuine domperidone on line. Regulation of fetal iron stores and its poor regulation in neonatal hemochromatosis associated with gestational alloimmune disease the fetus regulates placental iron transport to insure adequate iron for the growth and oxygen-carrying capacity needs of the fetus and newborn. Many of the control mechanisms that function after birth to control accrual of dietary iron also function during fetal life, with the placental trophoblast functioning in analogy to the duodenal mucosa (reviewed by Whitington and Kelly [36]). Ferroportin expression increases with gestational age in parallel with increasing iron needs of the fetus. Fetal hepcidin evidently regulates fetal iron stores: transgenic hepcidin-overexpressing mice are born profoundly anemic and iron deficient. This would be expected to limit the capacity for regulating placental iron flux and result in fetal iron overload. This results in high iron saturation and probably excess circulating non-transferrin-bound iron for uptake into tissues. Loss of hepatocytes is profound: hepatocyte volume density is usually less than 10% of normal newborn liver [40]. Residual hepatocytes appear damaged and may exhibit giant cell or pseudoacinar transformation. Portal triads are left intact, although hepatocyte loss and parenchymal collapse lead to their being crowded together. The hypercellular appearance in these patients comes from the presence of large numbers of oval cells, presumably from attempted regeneration, and in some cases macrophages. Maternal IgG binding to fetal hepatocytes activates fetal complement, with hepatocyte injury resulting from membrane attack complex. At autopsy, the most consistently affected tissues and the ones that should be most carefully examined with Perl Prussian blue staining are the exocrine pancreas, the thyroid follicles, the adrenal cortex, and the myocardium. Others in which siderosis can be found include epithelia of renal tubules, gastric and Brunner glands, parathyroid glands, and the thymus (Hassall corpuscles), as well as pancreatic islets, the adenohypophysis, and chondrocytes in hyaline cartilage. The spleen, lymph nodes, bone marrow, and other reticuloendothelial elements contain little or no stainable iron. In living patients, oral mucosal biopsy provides a mechanism of demonstrating extrahepatic siderosis, which can be found in the minor salivary glands. Correlation with the process of normal renal development dates arrested tubulogenesis to about 24-weeks of gestation. The liver is the source of angiotensinogen, which is required for development of proximal renal tubules. Residual hepatocytes take the form of pseudorosettes and multinucleate cells (trichrome stain, original magnification ױ00). The individual hepatocytes are condensed leaving large spaces between cords, which are filled with blood elements (trichrome stain, original magnification ױ00). Demonstration of extrahepatic siderosis is currently necessary to prove the diagnosis. No other disease of the newborn demonstrates the combination of severe liver disease and extrahepatic siderosis, and thus the combination of findings is absolutely diagnostic. The normal newborn liver contains sufficient stainable iron to be confused with pathologic siderosis, although they are qualitatively different to the eyes of experienced pathologists. Furthermore, pathologic hepatic siderosis has been described in several neonatal liver diseases. Also an important historical finding is one or more maternal siblings with early neonatal liver disease or death. The death need not be recorded as caused by liver failure since many of these cases are misdiagnosed as "sepsis" or carry non-specific diagnoses such as anasarca, hydrops, and bleeding diathesis. In some cases, the liver disease takes a prolonged course and is manifest days to weeks after birth. Twins may have disparate clinical findings, with one severely affected and the other minimally so [43]. Hypoglycemia, marked coagulopathy, hypoalbuminemia, edema with or without ascites, and oliguria are prominent features. Most infants exhibit significant elevations of both conjugated and non-conjugated bilirubin, with total bilirubin levels often exceeding 30 mg/dL. Serum aminotransferase concentrations are disproportionately low for the degree of hepatic injury, whereas circulating concentrations of -fetoprotein are characteristically very high, usually 100Ͷ00 g/mL (normal newborn values <80 g/mL). Studies of iron status often show hypersaturation of available transferrin, with hypotransferrinemia and hyperferritinemia (values >0. This child fully recovered with medical treatment (double volume exchange transfusion and intravenous immunoglobulin). The results of urine mass spectroscopy (see Chapter 21) do not show the findings characteristic of synthetic defects and usually show elevated levels of normal bile acids. Patients may receive the erroneous diagnosis of tyrosinemia based upon elevated serum tyrosine levels, which are reflective of failed hepatic metabolic function. Viral infections such as herpes simplex virus and echovirus must be excluded by appropriate examinations. Hemophagocytic lymphohistiocytosis also appears as a cause of neonatal liver failure. Biopsy of the oral mucosa is a clinically useful approach to obtain glandular tissue in which to demonstrate siderosis [44]. The two examinations are not always positive in the same patient, however, and together they have a sensitivity approaching 80%. Therefore, the diagnostic approach should be to perform one test and, if negative, do the other. The choice of which to start with is often determined by the ease with which it can be performed in the individual patient setting. Oral mucosal biopsy often fails because of an inadequate specimen not containing submucosal glands is obtained. Therefore, the surgeon should be instructed to take as deep and generous a specimen as possible. Examination of liver specimens for alloimmune injury involves immunohistochemistry for C5b-9 complex in hepatocytes [38]. This test is performed on paraffin-embedded archival tissue and so can be applied to autopsy materials from maternal siblings in order to achieve a diagnosis of a current patient. This test is at present only performed in one laboratory and is not commercially available. However, the question is asked as to whether the two approaches might be included in a therapeutic strategy. When it becomes clear that medical therapy has failed, usually in association with a catastrophic event, transplantation is no longer an option. Conversely, if transplant is performed, the potential for recovery with medical therapy is eliminated. This therapy was based on the hypothesis that oxidative injury caused by iron overload was central to disease pathogenesis, which appears not to be the case. The therapy can only ease ongoing alloimmune injury and permit recovery, which can take weeks to months. The infant must be supported in an intensive care setting with intravenous glucose infusions, blood products, and other therapies to prop up an ailing liver in order for recovery to occur. Sepsis and other catastrophic events may intervene and are the major reasons for the 20% failure rate of this therapy. Of the elements of the chelation antioxidant cocktail that might be retained in this therapy, only N-acetylcysteine and vitamin E have potential value outweighing negligible risk. Desferrioxamine should be avoided since it is toxic to neutrophils and its use incurs sepsis risk. With double-volume exchange transfusion and intravenous immunoglobulin therapy, recovery from liver failure to the point at which the infant can be discharged to home has varied from 1 to 4 months. Two such babies who are siblings underwent liver biopsy as neonates and again after 2ʹ years [48]. The current recommended treatment consists of intravenous immunoglobulin 1 g/kg body weight administered at 14 weeks, 16 weeks, and then weekly for a total of 20 doses.
Buy discount domperidone 10mg online
Scoliosis and kyphosis occur in approximately 50% of patients with Proteus syndrome (60 medicine vending machine domperidone 10 mg cheap, 66, 68, 69). Most of these patients who have progressive deformity do not respond to bracing and require spinal fusion. Other spinal deformities include localized spinal overgrowth and infiltration of the spinal canal by angiolipomatous tissue, which can cause both compression of the spinal cord and potential paraplegia (67, 70ͷ3). Other skeletal manifestations that have been found are hip dysplasia, exostoses that can limit joint movement, hindfoot deformity, and bony protuberances in the skull (61). Adolescent boy with Proteus syndrome with typical gyriform creasing of the sole of his foot. Adolescent boy with Proteus syndrome with recurrent left genu valgum after a high tibial osteotomy, just prior to repeat osteotomy. Splenomegaly and nephromegaly can occur along with various abnormalities in the brain (asymmetric megalencephaly and white matter changes) (53, 74, 75). Unusual tumors like ovarian cystadenomas, parotid adenomas, meningiomas, and others have been described but occur rarely (61, 76, 77). Thromboembolism is more common than in other syndromes with vascular malformations and can lead to sudden death even in children (78, 79). Orthopaedic surgery in patients with Proteus syndrome involves correction of leg length inequality and angular deformity and addressing macrodactyly. The leg length inequality is difficult to predict due to the abnormal growth patterns. Serial clinical and radiologic assessments need to be made to optimize the timing of the epiphysiodesis. When assessing the child with Proteus syndrome for surgery, the anesthetic and surgical team must be cognizant of the associated conditions that may affect surgery. The cystic lung disease, tonsillar hyperplasia, and increased risk of pulmonary embolus may affect the child (80). The first case of spontaneous absorption of bone was reported back in 1838; however, Gorham and Stout (81) and then Gorham (82) described the clinicopathologic features of the disease in the 1950s. Gorham disease usually occurs in the second or third decade of life; however, case reports have occurred from the neonatal period through to 65 years of age (835). There is no familial inheritance pattern of Gorham disease, and there is no greater incidence in either sex or any particular race. The most commonly involved bones are the maxilla, shoulder girdle, and pelvis although any bone can be involved including the spine (869). It can be recognized on x-rays as an incidental finding following trauma to an area. Occasionally, there is a history of pain that is not usually severe in the area of underlying osteolysis, and a child can present with some mild deformity of the involved area and some muscle weakness. On rare occasions, the patient may present with symptoms of a chylothorax where the lymphangiomatosis tissue has extended in to the chest from either shoulder girdle or spine involvement. Usually, the disease starts in one bone and can either remain there or spread to adjacent bones with complete disregard for the intervening joint or disc space (91). Other authors have shown that Gorham disease can arise in a multicentric pattern where more than one bone is initially involved but the intervening bone between the lesions is free of disease (92, 93). Although other causes of osteolytic lesions such as osteomyelitis and metastatic disease can be excluded on the clinical Gorham Disease. Gorham disease, sometimes referred to as "disappearing bone disease," is a rare condition that results in massive osteolysis of bone. A 13-year-old girl with Gorham disease who presented with a large right-sided pleural effusion. The plain radiographs will reveal either the monostotic involvement or multiostotic disease outlined above. The initial radiographic features were described by Johnson and McClure in 1958 (96). They show that initially there were multiple intramedullary and subcortical radiolucent foci with associated osteoporosis. They then describe an extraosseous stage when the cortex has been disrupted and there is an extension of the pathologic tissue into the adjacent soft tissues. The ends of the tubular bones then taper off to the area of osteolysis and this is thought to be due to compression by the surrounding soft-tissue involvement. One of the characteristic findings, however, is the lack of sclerosis or osteoblastic reaction in the area (97). Initially, with the neovascularization, there will be increased uptake on the T1 and T2 imaging; however, as the vascular tissue is replaced with fibrous tissue, the T1 and T2 imaging will become increasingly dark (86, 98, 99). Arteriography, venography, and lymphangiography have all been used to help in establishing a diagnosis and investigating the extent of the disease (83, 84, 97). Histologic slide of a biopsy in Gorham disease showing the thin-walled vessels lined by endothelium cells (arrow) and proteinaceous fluid. Histologic examination demonstrates benign endothelial proliferation within the bone. Some authors have reported the presence of osteoclasts in pathologic specimens (102ͱ04), whereas others have not found any to be present (87, 91, 97, 105). One of the mysteries of Gorham disease is the actual cause of this massive osteolysis. One thought is that the perivascular cells show some characteristics of osteoclast precursors that may form active osteoclasts (87, 106). Other authors have hypothesized that the osteoclast precursors in the area of osteolysis are more sensitive to humeral factors that promote osteoclast formation and bone reabsorption rather than an actual increase in actual osteoclast numbers (107, 108). With no obvious known cause and with such a variable natural history, the treatment of Gorham disease is extremely difficult. Although clinical and radiologic features usually confirm the diagnosis, a biopsy is often performed. The results of treatment have been particularly difficult to evaluate due to the variable natural history of Gorham disease. Occasionally, the multiple treatment modalities have been used over the years with variable results and still no consensus exists on the most effective treatment strategy. Guitierez and Spjut reviewed 25 patients with extraosseous involvement and found the mortality rate was higher in this group of patients with Gorham disease (105). The extraosseous tissue can invade the pleural cavity giving rise to pulmonary complications due to the persistent chylothorax. This can occur when the shoulder girdle is involved or the thoracic or lumbar spine (90, 91, 109). The management of the chylothorax is extremely difficult and usually not successful. Attempts have been made to tie off the thoracic trunk and use different radiation and pleural adhesion therapies (86). For more straight forward monostotic disease, the treatment has also been variable and disappointing. Spontaneous regression without treatment can occur; however, in the majority of cases, no new bone is ever made. Local excision of the affected bone and soft tissue and bone grafting with or without internal fixation has also been largely unsuccessful (89, 110, 111). Unfortunately, most of the bone grafts are reabsorbed, and this has encouraged some authors to use vascularized fibula grafts to strut the affected area (112). The use of an endoprosthesis to bridge the gap between the so-called "normal bone" can be successful (85, 89, 112, 113). When limb salvage is not possible due to the extent of both the bone and the soft-tissue disease, then amputation is often indicated. In a limb with extensive bone disease and massive soft-tissue involvement, it may be more functional for the patient to have an amputation or limb disarticulation rather than a salvage procedure. Predicting the level of the amputation is difficult as through joint amputation has not always been shown to control the disease process (93, 98, 114). One can see that not only the type of surgical procedure is a challenge to decide but the timing of the surgical intervention may also be critical. In rapidly evolving disease, it is difficult to know whether to intervene "early" to try and halt the progress or wait until the disease process has slowed down to assess the extent of the reconstruction or amputation. A pathologic fracture through the area of osteolysis has been shown to increase the rapidity of the osteolytic process and Mendez et al. The management of vertebral Gorham disease is extremely challenging as resection with adequate margins is usually not possible. Spinal bracing is a temporizing treatment; however, definitive surgical intervention is required when there is neural compromise.
Order discount domperidone line
The Q angle is measured with the knee in 30 degrees of flexion so that the patella is in contact with the femoral sulcus symptoms questions purchase domperidone without a prescription. Patients with an increased Q angle (>15 degrees) may have knee pain because of lateral tracking of the patella in the femoral sulcus resulting in a small contact area between the patella and femur. The maltracking can be detected by observing the patella as the patient actively extends the knee. Sitting over the side of the table with the knees flexed to 90 degrees, the patient is asked to gradually extend the knee. The patella is observed to remain in the femoral sulcus as it ascends along the axis of the femur, but as the knee reaches full extension, the patella deviates laterally like an upside-down J. This is termed a positive "J sign," and if the patient has patellofemoral instability, the patella may subluxate with this maneuver. It is important to note that a normal knee has a "J sign," but in an unstable knee it is exaggerated or occurs earlier or more dramatically compared to the normal side as the knee extends. A patient with anterior knee pain will typically have a full range of motion from 0 to 135 degrees. The pain is often described as being circumferential around the patella, and there is usually no evidence of an effusion. If the lesion is in the lateral aspect of the medial femoral condyle, the pain can be reproduced by flexing the knee to 90 degrees, internally rotating the tibia, then gradually extending the knee. A torn meniscus can be evaluated by maximally flexing the knee and circumducting the tibia on the femur. If the clinician palpates a clunk with this maneuver, a meniscus tear is likely and this is termed a positive "McMurray test. With the patient in the prone position, this test is performed by applying pressure directly to the heel, loading the knee in compression, while the tibia is internally and externally rotated. A patient with a torn meniscus will experience pain with this maneuver when the meniscus gets trapped between the tibia and the femur. With the patient supine and the knee extended, the clinician elevates the foot, then drops it several inches causing the knee to hyperextend, flex, then hyperextend again. Most patients will not have discomfort with this maneuver, but if there is a torn meniscus the maneuver causes pain at the medial joint line and a reflex contraction of the hamstrings preventing the knee from hyperextending. This inability to hyperextend the knee associated with medial joint line pain is termed a positive "bounce test. This test is performed with the patient in the supine position and the knee flexed to 90 degrees with the foot flat on the exam table. The patient is asked to slide her foot directly down the table by contracting the quadriceps muscles, while the clinician prevents the foot from moving. The quadriceps active test is performed with the patient supine and the knee flexed to 90 degrees. The patient is asked to slide her foot down the table, while the clinician prevents the foot from moving (down arrow). The force of the quadriceps muscle pulls the tibia anteriorly (up arrow), reducing the posterior subluxation. This test is performed with the patient supine and the knee flexed to 90 degrees with the foot flat on the exam table. A valgus and internal rotation force is applied to the lateral tibia while the calcaneus is grasped with the other hand. As the knee is flexed, when the iliotibial band crosses the axis of the knee joint, the tibia rapidly shifts to its normal position, and a pivot shift or jerk is felt by the clinician and the patient. This test can only be reliably performed when the patient is completely relaxed, so it is usually not helpful in an acutely injured patient. The anterior drawer test is performed with the patient supine and the knee flexed to 90 degrees. The medial and lateral collateral ligaments are located just under the skin, so an injury to these ligaments is associated with pain to palpation over the ligament. The Lachman test is performed with the patient supine and the knee flexed to 30 degrees. To test the left knee, the femoral condyles are held with the right hand, while the tibia is pulled anteriorly with the left hand (arrow). The medial and lateral collateral ligaments are tested with the knee in 30 degrees of flexion, because varus or valgus instability can be masked by intact cruciate ligaments with the knee in extension. The medial joint line is palpated with a finger, while the examiner applies a valgus stress to the knee, and the lateral joint line is similarly palpated while the examiner applies a varus stress to the knee. The amount of joint line opening is recorded in millimeters, and 0 to 5 mm of opening with a solid end point is considered a normal amount of ligamentous laxity. Medial and lateral collateral ligament sprains are classified according to the amount of opening of the joint space on physical examination. This patient has anterior knee pain or patellofemoral pain secondary to a direct blow to the patella when she fell on the knee. Activity modification and physical therapy is recommended in anticipation of gradual improvement. The last episode occurred 1 month ago, when he was walking downstairs and the left ankle gave out, causing swelling over the lateral aspect of the ankle. The pain under both arches has worsened over the last year and is aggravated by exercise. The clinician understands that pes cavus is associated with a neuromuscular disorder until proven otherwise. The most common neurologic disorder associated with pes cavus is Charcot-Marie-tooth disease. The inheritance pattern for Charcot-Marie-tooth disease is autosomal dominant, so the clinician suspects that this boy may have Charcot-Marie-tooth disease, since his father and grandfather both have high arches. On physical examination, he ambulates with a heel-toe-gait pattern, but walks on the lateral side of the foot with the heel in varus. In standing, he has high-arched feet and points to the medial arch of both feet when describing the pain. He has painful callosities on both feet under the heels, the first and fifth metatarsals, over the dorsal surfaces of the proximal interphalangeal joints of the lateral toes. A detailed motor, sensory, and reflex examination of the upper and lower extremities is within normal limits. The longitudinal arch of each foot is elevated, shortening the medial border of the foot, creating a concave appearance. The lateral side of each foot is convex, with lengthening of the lateral border of the foot, creating a bean-shaped deformity. On both feet, the first metatarsal is plantarflexed, forcing the hindfoot to rotate into a varus position. The normal foot has a "tripod" structure with weight bearing balanced between the heel, the first metatarsal head, and the fifth metatarsal head. If the forefoot develops a pronation deformity, with plantarflexion of the first metatarsal, weight bearing will force the hindfoot into varus. The flexible hindfoot varus deformity will eventually become a structural deformity as the soft tissues of the subtalar joint contract over time. The flexibility of the hindfoot deformity is important when contemplating surgical reconstruction of a cavus foot. The forefoot contribution to the hindfoot varus deformity is determined by the Coleman block test (43). The amount of correction of the hindfoot deformity, when standing on the block with the first metatarsal off the medial side of the block, represents the forefoot contribution to the hindfoot varus deformity. A similar test can be performed with the patient prone and the knee flexed to 90 degrees. The foot is dorsiflexed by applying pressure over the fifth metatarsal head, allowing the first metatarsal to remain plantarflexed, and the amount of correction of the hindfoot varus is observed. These tests are crucial in preoperative planning because a flexible hindfoot will correct when the forefoot deformity is corrected, whereas a rigid hindfoot will not. This patient has pes cavus that may be associated with Charcot-Marietooth disease, so a referral to a geneticist is recommended. In addition, standing anteroposterior, lateral, oblique, and Harris axial radiographs of the calcaneus are recommended.
Immunoglobulin IgY (Hyperimmune Egg). Domperidone.
- How does Hyperimmune Egg work?
- Rotaviral diarrhea, infectious diarrhea, arthritis including osteoarthritis and rheumatoid arthritis, and high cholesterol.
- What is Hyperimmune Egg?
- Dosing considerations for Hyperimmune Egg.
- Are there safety concerns?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=97074
Discount 10 mg domperidone mastercard
Infantile apnoea due to profound hypocalcaemia associated with vitamin D deficiency symptoms vaginitis generic 10 mg domperidone with amex. Asian osteomalacia is determined by dietary factors when exposure to ultraviolet radiation is restricted: a risk factor model. The importance of limited exposure to ultraviolet radiation and dietary factors in the aetiology of Asian rickets: a risk-factor model. Pathophysiology of calcium metabolism in children with vitamin D-deficiency rickets. Single-day therapy for nutritional vitamin D-deficiency rickets: a preferred method. Vitamin D-resistant rickets: analysis of twenty-four pedigrees with hereditary and sporadic cases. Prevalence of dental abscess in a population of children with vitamin D-resistant rickets. New perspectives on the biology and treatment of X-linked hypophosphatemic rickets. The effect of osteotomy on bowing and height in children with X-linked hypophosphatemia. The effect of treatment on growth and deformity in hypophosphatemic vitamin D-resistant rickets. Clinical implications of genetic defects in G proteins: the molecular basis of McCuneAlbright syndrome and Albright hereditary osteodystrophy. Eine besondere form des primare vitamin D resistenten rachitis mit hypocalcemie und autosomal-dominanten erbgang: Die hereditare pseudomangelrachitis. Cloning of human 25-hydroxyvitamin D-1 alpha-hydroxylase and mutations causing vitamin D-dependent rickets type 1. A clinical and research protocol for characterizing patients with hypophosphatasia. Bilateral slipped upper femoral epiphysis: a rare manifestation of renal osteodystrophy. Slipped capital femoral epiphyses complicating renal osteodystrophy: a report of three cases. Does inadequate diet during childhood explain the higher high fracture rates in the Southern United States? Self-reported lifetime physical activity and areal bone mineral density in healthy postmenopausal women: the importance of teenage activity. Weight-bearing exercise and bone mineral accrual in children and adolescents: a review of controlled trials. Osteogenesis imperfecta: a study of clinical features and heredity based on 55 Danish families comprising 180 affected persons. Osteogenesis imperfecta: a clinical and biochemical study of a generalized connective tissue disorder. Is it necessary to screen for hearing loss in the paediatric population with osteogenesis imperfecta? Displaced fractures of the apophysis of the olecranon in children who have osteogenesis imperfecta. Type I osteogenesis imperfecta: a nonfunctional allele for pro alpha (I) chains for type I procollagen. The role of dual energy x-ray absorptiometry in aiding the diagnosis of pediatric osteogenesis imperfecta. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. Skeletal effects and functional outcome with olpadronate in children with osteogenesis imperfecta: a 2-year randomised placebo-controlled study. Vertebral morphometry in children and adolescents with osteogenesis imperfecta: effect of intravenous pamidronate treatment. Effect of intravenous pamidronate therapy on everyday activities in children with osteogenesis imperfecta. Long-bone changes after pamidronate discontinuation in children and adolescents with osteogenesis imperfecta. Pamidronate does not adversely affect bone intrinsic material properties in children with osteogenesis imperfecta. Bone healing in children with osteogenesis imperfecta treated with bisphosphonates. The outcome of pregnancy following pre-pregnancy or early pregnancy alendronate treatment. No osteonecrosis in jaws of young patients with osteogenesis imperfecta treated with bisphosphonates. Alendronate affects long bone length and growth plate morphology in the oim mouse model for osteogenesis imperfecta. Effects of N,N,NԬNեthylenediaminetetramethylene phosphonic acid and 1-hydroxyethylidene1,1-bisphosphonic acid on calcium absorption, plasma calcium, longitudinal bone growth, and bone histology in the growing rat. Complications of intramedullary rods in osteogenesis imperfecta: Bailey-Dubow rods versus nonelongating rods. Functional results of operation in osteogenesis imperfecta: elongating and nonelongating rods. Osteogenesis imperfecta: treatment bymultiple osteotomy and intramedullary rod insertion. Management of lower-extremity deformities in osteogenesis imperfecta with extensible intramedullary rod technique: a 20-year experience. Osteogenesis imperfecta: profiles of motor development as assessed by a postal questionnaire. Osteogenesis imperfecta in childhood: impairment and disability - a follow-up study. Scoliosis in children with osteogenesis imperfecta: influence of severity of disease and age of reaching motor milestones. Operative treatment of severe scoliosis in osteogenesis imperfecta: results of 20 patients after halo traction and posterior spondylodesis with instrumentation. Spinal deformity, pulmonary compromise, and quality of life in osteogenesis imperfecta. Radiographic classification, natural history, and treatment of spinal deformities. Correlation of scoliotic curvature with Z-score bone mineral density and body mass index in patients with osteogenesis imperfecta. Prevalence of vertebral pars defects (spondylolysis) in a population with osteogenesis imperfecta. Surgical management of severe cervical kyphosis with myelopathy in osteogenesis imperfecta: a case report. Bruck syndrome: a rare combination of bone fragility and multiple congenital joint contractures. Defective collagen crosslinking in bone, but not in ligament or cartilage, in Bruck syndrome: indications for a bone-specific telopeptide lysyl hydroxylase on chromosome 17. Osteoporosis-pseudoglioma syndrome: report of three affected sibs and an overview. Osteoporosis pseudoglioma syndrome: treatment of spinal osteoporosis with intravenous bisphosphonates. Failure of operative treatment in a child with osteoporosis-pseudoglioma syndrome. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Bone density and metabolism in children and adolescents with moderate to severe cerebral palsy. Changes in skeletal maturation and mineralization in children with cerebral palsy and evaluation of related factors. Bone mineralization in the affected extremities of children with spastic hemiplegia.
Syndromes
- Urine pH
- Heart failure
- Ultrasound of the abdomen or pelvis
- Duodenal ulcer
- Phoratoxin
- If you are or could be pregnant
- Alcoholic liver disease
- Joint or muscle pain
- Eat a heart-healthy diet, exercise, stop smoking (if you smoke), and reduce stress to help lower your chances of having a blocked artery again.
- DO NOT try to make the person stop convulsing. He or she has no control over the seizure and is not aware of what is happening at the time.
Trusted domperidone 10mg
Incidence may be higher in specific groups symptoms zollinger ellison syndrome best purchase domperidone, including residents of group homes, male homosexuals, and immigrants from endemic areas. After traversing the stomach, ingested cysts dissolve during passage through the small bowel and colon where, in the presence of colonic bacteria, they mature into trophozoites. Colonic infection may be asymptomatic or may manifest as invasive disease characterized by abdominal pain, bloody diarrhea, and the presence of "pipe stem" ulcers. Subsequent spread of trophozoites to other organs, including brain, lung, heart, and spleen, has been described. In addition, local spread of amebic organisms may result in the presence of cutaneous ulcerations, most commonly noted in the perineal area in children. Hepatic abscess formation is estimated to occur in 1ͷ% of children with invasive amebiasis. Children under 3 years of age seem to be most commonly affected; no male:female differential in incidence exists in this age group. Single or multiple cavities may be present; the right lobe of the liver is most commonly involved. The abscess cavity consists of a liquefied central area, surrounded by necrotic hepatic tissue. The trophozoites, which are oval eosinophilic organisms, contain a single nucleus ranging in size from 10 to 65 m. The cytoplasm of the trophozoites is characteristically granular and vacuolated, and often contains phagocytosed erythrocytes. Signs and symptoms of amebic abscess include fever, abdominal pain, abdominal distension, and tender hepatomegaly, but presentation in young children may be non-specific. Patients may present with an acute abdomen secondary to intraperitoneal abscess rupture. Free perforation into the peritoneal cavity is, however, less likely than slow leakage with intra-abdominal abscess formation. Other reported symptoms include dyspnea and productive cough, occasionally as a result of abscess rupture into the chest with formation of a hepatobronchial fistula. Patients in whom an intrahepatic amebic abscess has ruptured into the pericardium may present in shock. History of a preceding diarrheal episode is present in less than 50% of patients; approximately 10% have dysentery concurrent with hepatic abscess. Routine laboratory examinations are of limited value in the diagnosis of hepatic amebic abscess. Serum aminotransferases are elevated in less than 25% of affected children, and serum alkaline phosphatase is generally normal in this age group [59]. Radiography of the chest may reveal right lower lobe infiltrates, pleural effusions, or elevation of the right or left hemidiaphragms. Hepatic scintigraphy has proven useful in adult series; a filling defect is noted corresponding to abscess location. Examination of stools for trophozoites or cysts should be performed but is positive in less than 50% of patients with hepatic amebic abscess. Serologic testing for ameba is a useful diagnostic tool, particularly in areas which have relatively low incidences of amebic disease. Approximately 95% of patients with amebic liver abscess have positive enzyme immunoassays [59]. These studies remain positive for significant periods of time after acute infection. As a means of differentiation from pyogenic abscess, fine-needle aspiration of the abscess cavity is often required. Examination of this "pus" yields amebic organisms in less than one-third of patients. Therapy of amebic abscess consists primarily of the use of amebicidal agents, most often metronidazole 50 mg/kg per day in divided doses for 10 days, followed by a luminal amebicide such as iodoquinol or paromomycin. Nitazoxanide has been shown efficacious as a single agent in preliminary studies [60]. Surgical drainage is generally not required and mortality rates appear to be significantly higher when this is employed. Indications for surgical drainage include presentation with an acute abdomen as well as failure of other therapeutic measures. Percutaneous aspiration is more frequently utilized, although evidence of added efficacy to medical therapy alone is unclear [61]. Current indications include poor response to amebecidal therapy after 4͵ days, or as decompressive therapy in those patients in whom rupture of the abscess cavity, into the pleural, peritoneal, or pericardial cavities, seems imminent. Rupture of the abscess cavity is associated with significant mortality, approximately 30% die when rupture into the pericardium occurs. Conversely, mortality rates in adults with recognized, uncomplicated, hepatic amebic abscess are approximately 1%. Current pediatric mortality rates are unclear but may be significantly higher because of delays in diagnosis. Echinococcal disease Human infection with Echinococcus granulosus may occur after ingestion of ova excreted by infected dogs. Dogs generally acquire infection via consumption of sheep liver and/or intestine containing hydatid cysts. Scolices contained within the cysts then develop within the canine small intestine, maturing into adult tapeworms, 3Ͷ mm long. Rupture of the gravid proglottid releases 40000 eggs, which are excreted in canine feces. Ingestion of eggs typically occurs after the handling of an infected dog or the drinking of contaminated water. The ingested embryo, after release from the egg in the duodenum, penetrates the intestinal mucosa and enters the portal circulation. Although in adult series involvement of the liver occurs three times more frequently than the lung, involvement of the lung is noted frequently in children. Other sites of infection in approximately 10% of children include the brain, bones, genitourinary tract, eyes, spleen, and heart. Hepatic involvement is marked by the development of "cysts" within the hepatic parenchyma, most often within the right lobe. Hydatid sand, composed of separated brood cysts and protoscolices, floats within the main cyst cavity. Symptoms of echinococcal cyst formation occur as a result of cyst growth and subsequent compression of surrounding tissues. Therefore, right upper quadrant pain and fullness may be the only presenting features. Jaundice may occur as a result of compression of the porta hepatis; cholangitis may arise secondary to cyst rupture into the biliary tract. Laboratory data are typically non-specific; elevation of serum alkaline phosphatase and aminotransferases may be noted. Radiography of the abdomen may show calcification of the cyst wall in adults; this change is seldom apparent in children. Ultrasound is useful and can demonstrate the presence of hydatid sand as well as delineate septations and the presence of daughter cysts. Endoscopic retrograde cholangiography may demonstrate involvement of the biliary tree by daughter cysts following rupture of the primary hepatic cyst. Large lesions require may still require cyst decompression, irrigation with scolicidal solutions, and, in some cases, omentoplasty [64]. Laparoscopic approaches have also been utilized successfully; care is taken to avoid peritoneal dissemination at surgery. Daughter cysts in the biliary tree may be removed endoscopically after sphincterotomy [65]. In unapproachable hepatic lesions, therapy with albendazole, 15 mg/kg daily for 1 to 6 months, has in some cases resulted in reduction in cyst size. Ascariasis Human infection with the roundworm Ascaris lumbricoides is extremely common in tropical and temperate regions worldwide.
Buy domperidone 10mg
They may have shortening of the first metatarsal with hyperextension of the metatarsophalangeal joint reflecting a plantarflexed first ray medicine in the middle ages buy domperidone online pills. If these reflexes persist beyond 10 months of age, it may be a sign of a neuromuscular disorder. The neck-righting reflex is elicited by turning the head to one side; it is positive if the trunk and limbs spontaneously turn toward the same side. The Moro reflex is elicited by gently lifting the infant with the right hand under the upper thoracic spine and the left hand under the head. The infant abducts the upper limbs, with spreading of the fingers, followed by an embrace. Similarly, extension of the neck causes extension of the upper limbs and flexion of the lower limbs. The asymmetric tonic neck reflex is elicited by turning the head to the side, which causes extension of the upper and lower extremities on the side toward which the head is turned, and flexion of the upper and lower extremities on the opposite side. The extensor thrust, an abnormal reflex, is elicited by holding the infant under the arms and touching the feet to the floor, which causes a rapid extension of all of the joints of the lower limb, progressing from the feet to the trunk. A normal infant will flex rather than extend the joints of the lower extremities when placed in this position. These primitive reflexes need to resolve with growth and development before the child will be able to walk independently. There are other primitive reflexes that gradually disappear in normal children at different stages of development, including the rooting, startle, Gallant, and Landau reflexes. The rooting reflex is elicited by touching the corner of the mouth, which causes the mouth and tongue to turn toward the side that was stimulated. The startle reflex is elicited by making a loud noise, which causes a mass myoclonic response resembling a Moro reflex, except that the elbows remain flexed. The Gallant reflex is elicited by stroking the side of the trunk, which causes the infant to bend the spine toward the side that was stimulated, creating a scoliosis convex to the opposite side that was stimulated. The Landau reflex is elicited by supporting the infant by the trunk in the horizontal prone position; the typical response is extension of the neck, spine, and extremities. The reflex is positive if the infant extends the upper extremities to break the fall. The footplacement reaction is elicited by holding the infant under the arms, then gently lifting the infant so that the dorsum of the foot or the anterior surface of the tibia touches the side of the table. The foot-placement reaction usually develops early in infancy and may persist until the age of 3 or 4 years. Bleck (12) evaluated 73 children who were 12 months of age or older and were still not yet walking to determine their prognosis for walking. One point was assigned for each primitive reflex that was still present, and one point was assigned for each postural reflex that was still absent (Table 4-4). A score of two points or more indicated a poor prognosis for walking, a one-point score indicated a guarded prognosis, and a zero-point score indicated a good prognosis. The physical examination continues by evaluating the spine for any scoliosis or kyphosis. The upper and lower extremities are examined to assess range of motion and to document any contractures. If a contracture is identified, the clinician attempts to passively correct it to determine if it is flexible or rigid. The foot-placement reaction is elicited by gently lifting the infant so that the dorsum of the foot or the anterior surface of the tibia touches the side of the table. When the clinician gradually attempts to passively correct a rigid contracture, if the contracture has continuous resistance to passive correction, it is termed "leadpipe rigidity. The parachute reflex is elicited by holding the infant in the air in the prone position, then suddenly lowering the infant headfirst toward the table, simulating a fall. The reflex is positive if the infant extends the upper extremities as if to break the fall (arrow). Primitive reflex Asymmetric tonic neck Neck righting Moro Symmetric tonic neck Extensor thrust Postural reflex Parachute Foot placement Prognosis for walking: 2 points, poor; 1 point, guarded (might walk); 0 points, good. If the athetosis is of the tension type, it can often be "shaken out" of the limb by the clinician. The reflexes are also tested to determine if the patient has hyperreflexia, clonus, and a positive Babinski reflex. A 3-Month-Old Boy Is Referred for Evaluation Because He Is Not Moving His Right Arm. Shortly after delivering a healthy 5250 g (11 lb 9 oz) baby boy, the mother was told that the baby was not moving his right arm. The pregnancy was normal, but the delivery was difficult because of right shoulder dystocia. The delivery team had to apply considerable traction on the head to deliver the baby. At the 2-month appointment with the pediatrician, he was moving his hand but always kept the upper extremity at his side. After a pediatric orthopaedic history and physical examination, the clinician focuses on a detailed examination of the upper extremities, comparing the paralyzed right side with the uninvolved side. It is important to distinguish between a brachial plexus palsy (a traumatic paralysis involving the upper extremity) and a pseudoparalysis secondary to osteomyelitis of the proximal humerus, septic arthritis of the shoulder, or a birth fracture. The treatment for each of these conditions is different, and a delay in treatment of osteomyelitis or septic arthritis can be devastating. An infant with osteomyelitis, septic arthritis, or a birth fracture will usually have swelling at the site, whereas an infant with traumatic brachial plexus palsy will have no swelling in the extremity, but may have swelling in the neck. An infant with a brachial plexus birth palsy or birth fracture of the humerus will usually have paralysis at birth, whereas an infant with osteomyelitis or septic arthritis may be normal after birth, and then suddenly develop the pseudoparalysis. Traumatic brachial plexus palsy is a common birth injury, typically seen in primigravida mothers with large babies after difficult deliveries. It occurs because of traction and lateral tilting of the head to deliver the shoulder. If the baby is in the breech presentation, it occurs because of traction and lateral tilting of the trunk and shoulders to deliver the head. Traumatic brachial plexus palsy may have an associated fracture of the clavicle or humerus. There are three types of brachial plexus palsies, depending on which part of the brachial plexus is affected. The Erb palsy affects the upper roots (C56), the Klumpke palsy affects the lower roots (C8 and T1), and total plexus palsy affects all of the roots in the brachial plexus. The prognosis for recovery depends on the level and magnitude of the injury and the time at which certain key muscles recover function. If the biceps recovers function before 3 months of age, the prognosis is excellent for a full recovery. Paralysis of C5 and C6 causes the shoulder to be held in adduction and internal rotation, with the elbow in extension, the forearm in pronation, and the wrist and fingers in flexion. The paralysis of C5 and C6 causes the shoulder to be held in adduction and internal rotation, with the elbow in extension, the forearm in pronation, and the wrist and fingers in flexion. This posture is not seen in an infant with osteomyelitis, septic arthritis, or birth fracture. A Horner syndrome refers to the constellation of signs resulting from the interruption of sympathetic innervations to the eye and ocular adnexae. The clinical findings include a triad of ipsilateral blepharoptosis, pupillary miosis, and facial anhidrosis. If the infant has a Horner syndrome, the prognosis for spontaneously recovery is decreased. This 3-month-old boy is already starting to show biceps motor function at 3 months of age, so the prognosis for recovery is good. An 18-Month-Old Boy Is Referred Because He Refuses to Walk After His Mother Fell While Carrying Him. When she tried to stand him up, he would not put any weight on his right lower extremity. She has not noticed any swelling and he stopped crying after she gave him some anti-inflammatory medication. The radiograph showed a fractured tibia so he referred them for evaluation and treatment. The age of the boy is important because most child abuse involves children younger than 3 years of age.
Purchase domperidone toronto
Aldolase B mutations in Italian families affected by hereditary fructose intolerance treatment modalities order domperidone 10 mg without prescription. Expression, purification, and characterization of natural mutants of human aldolase B. Mutations in the promoter region of the aldolase B gene that cause hereditary fructose intolerance. Absence of renal fructose-1-phosphate aldolase activity in hereditary fructose intolerance. A structurally modified aldolase in fructose intolerance: immunologic and kinetic evidence. Structural and functional analysis of aldolase B mutants related to hereditary fructose intolerance. Essential fructosuria, hereditary fructose intolerance, and fructose-1,6-diphosphatase deficiency. Diagnosis of fructose-1,6bisphosphatase deficiency using cultured lymphocyte fraction: a secure and noninvasive alternative to liver biopsy. Glykogenspeicherkrankheit der Leber und Nieren [Hepato-nephromegalia glykogenica]. Studies of liver glycogenoses, with particular reference to the metabolism of intravenously administered glycerol. The glucose-6-phosphate transporter is a phosphate-linked antiporter deficient in glycogen storage disease type Ib and Ic. Isolation of the gene for murine glucose-6phosphatase, the enzyme deficient in glycogen storage disease type 1A. Identification of mutations in the gene for glucose-6-phosphatase, the enzyme deficient in glycogen storage disease type 1a. The growth hormone-insulin-like growth factor axis in glycogen storage disease type 1: evidence of different growth patterns and insulin-like growth factor levels in patients with glycogen storage disease type 1a and 1b. Lactate as a cerebral metabolic fuel for glucose-6-phosphatase deficient children. Nocturnal intragastric therapy in type I glycogen storage disease: effect on hormonal and amino acid metabolism. Optimal rate of enteral glucose administration in children with glycogen storage disease type I. Studies of uric acid metabolism in glycogen storage disease associated with gouty arthritis. Excessive activity of the pathway in hypoxanthine-guanine phosphoribosyltransferase deficiency. Regression of hepatic adenomas in type Ia glycogen storage disease with dietary therapy. Natural history of hepatocellular adenoma formation in glycogen storage disease type 1. Hepatocellular glycogenosis and related pattern of enzymatic changes during hepatocarcinogenesis. Chromosomal and genetic alterations in human hepatocellular adenomas associated with type Ia glycogen storage disease. Portacaval shunt for glycogen storage disease: value of prolonged intravenous hyperalimentation before surgery. Comparison of the effects of total parenteral nutrition, continuous intragastric feeding, and portacaval shunt on a patient with type I glycogen storage disease. On the involvement of a glucose 6-phosphate transport system in the function of microsomal glucose 6-phosphatase. The role of the membrane in the regulation of activity of microsomal glucose-6phosphatase. Inactivation of the glucose 6-phosphate transporter causes glycogen storage disease type 1b. StructureΦunction analysis of the glucose-6-phosphate transporter deficient in glycogen storage disease type Ib. Clinical and biochemical findings before and after portacaval shunt in a girl with type Ib glycogen storage disease. Improvement of neutropenia and neutrophil dysfunction by granulocyte colony-stimulating factor in a patient with glycogen storage disease type Ib. Vitamin E supplementation improves neutropenia and reduces the frequency of infections in patients with glycogen storage disease type 1b. Glycogen storage disease: report of a case with abnormal glycogen structure in liver and skeletal muscle. Studies on glycogen disease with report of a case in which the glycogen was abnormal. Amylopectinosis disease isolated to the heart with normal glycogen branching enzyme activity and gene sequence. Sokol 28 Introduction the accumulation of excess copper in the liver is toxic in humans and other mammals and may lead to hepatitis, fulminant hepatic failure, cirrhosis, and death. The therapeutic success using oral copper-chelating agents and zinc therapy make Wilson disease one of the few treatable genetic metabolic liver diseases. In a fulminant presentation or advanced disease at diagnosis, copper chelation is ineffective and liver transplantation is life-saving. Copper absorption and metabolism the normal adult Western diet contains 2͵ mg per day of copper. The efficiency of copper absorption in adults ranges from 40 to 60% [1], with higher absorption at lower intakes. Foods containing high amounts of copper include unprocessed wheat, dried beans, peas, shellfish (particularly oysters), chocolate, liver, and kidney. For example, excess intake of zinc, cadmium, and ascorbic acid can interfere with copper bioavailability because of the formation of insoluble copper salts at an alkaline pH [3]. A vegetarian diet, as well as ingested raw meat, have been associated with decreased copper absorption. It is the balance between these exogenous and endogenous factors that regulate intestinal absorption of copper. Because dietary intake and absorption generally exceed metabolic needs, a large amount of ingested copper is eventually excreted in bile (see below). Although enterocyte metallothionein also binds zinc and cadmium, copper is bound most avidly. Because zinc stimulates metallothionein synthesis, it has been shown that increased dietary zinc may impair copper absorption by causing retention of copper in the enterocyte, which is then excreted in the feces following desquamation of the enterocyte. This mechanism forms the basis for oral zinc therapy in Wilson disease (see below). Once absorbed into the portal venous blood, copper is complexed to albumin and amino acids in equilibrium with a very small fraction of free ionic copper. Among the amino acids present in blood, the binding affinities for copper in decreasing order are histidine, threonine, glutamine, and asparagine. This amino acid-bound copper is most likely the form in which copper is transported to various tissues other than the liver. The function of these proteins is to store copper for subsequent metabolic needs of the hepatocyte, to bind and detoxify excess copper, and to provide copper to chaperones that assist in incorporating it into essential proteins that are secreted. Important hepatic copper metalloenzymes include superoxide dismutase (32 kDa), mitochondrial monoamine oxidase (195 kDa), cytochrome c oxidase (290 kDa), and ceruloplasmin. This process represents the main homeostatic mechanism for copper metabolism in humans. Copper conjugated to glutathione is a minor pathway of copper excretion into bile. Metallochaperones play an essential key role in copper homeostasis in mammalian (and yeast) cells. These proteins control delivery of copper to specific intracellular targets, in which copper is incorporated into synthesis of critical enzymes and proteins [7]. Ceruloplasmin, a blue-colored copper-containing 2globulin (134 kDa), is synthesized mainly by hepatocytes and secreted into the systemic circulation [10].
