

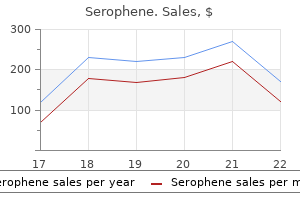
Proven 100mg serophene
Because the double-stranded form cannot be translated and is probably degraded rapidly womanlog pregnancy buy serophene overnight delivery, the production of its protein product is inhibited specifically. Once a cell in which recombination has occurred within a gene is found, it can be implanted into a blastocyst, with the hope that some of the progeny of the implanted cell will become germ cells. The value of the technique is often limited by the fact that the knockout may be lethal. But in some diseases, such as hemochromatosis,58 knockout models of various forms of the disease are valuable resources. In situations in which a knockout proves to be lethal, or where it would be useful to limit the deficiency to a single-organ system, the Cre/LoxP site-specific recombination system has proven to be very useful. Tissuespecific excision can be achieved by inserting the Cre-recombinase downstream from a tissue-specific promoter. For example, a mutation in a membrane serine protease of unknown function revealed that it was a negative regulator of hepcidin, and subsequent investigations revealed that mutations of this gene caused hereditary iron deficiency in humans. It is also possible to carry out germline therapy, which affects all cells, including reproductive cells, but for technical and ethical reasons this is not being pursued in humans. Most commonly, somatic cell therapy is used for conditions in which a mutation has caused the absence of a gene product in a cell. These vectors are usually viruses, such as retroviruses or adenoviruses, which have been genetically modified so that they contain the normal human gene and cannot make copies of themselves (otherwise they could cause a viral infection). In one case, an immune response against an adenoviral vector proved fatal, and several cases of leukemia have resulted from the insertion of a modified retrovirus near an oncogene. It is hoped that further research will lead to safe, efficient and cost-effective treatment of many human diseases through gene therapy. The use of promoters that are inducible or tissue specific permits studies of the effect of a gene product that might be lethal if expressed in all tissues or at all times during embryogenesis. Kayser M, de Knijff P: Improving human forensics through advances in genetics, genomics and molecular biology. Soda Y, Tani K, Bai Y, et al: A novel maxizyme vector targeting a bcr-abl fusion gene induced specific cell death in Philadelphia chromosome-positive acute lymphoblastic leukemia. Cavazzana-Calvo M, Fischer A, Hacein-Bey-Abina S, Aiuti A: Gene therapy for primary immunodeficiencies: Part 1. Mardis the introduction of next-generation sequencing platforms, coincident with genome-scale preparatory and analytical approaches and the completion of the Human Genome Reference, has ushered in the era of genomics. This chapter introduces the fundamentals of next-generation sequencing methods, provides an overview of the basics of data analysis, and explores the myriad applications developed to exploit the scale and throughput of next-generation sequencing toward questions of biomedical importance. Specifics of cancer genomics, complex disease genomics, and how they pertain to hematologic basic science and clinical practice are discussed, along with the modern-day realities of the consenting process. Frederick Sanger and his colleagues developed Sanger or "chain termination" sequencing in the late 1970s. The main limitation of Sanger sequencing is that the sequencing reaction is decoupled from the electrophoretic separation and detection steps. Currently, Sanger sequencing is still in use to complete smaller scale sequencing projects and to validate findings from next-generation sequencing studies. In general, this in situ amplification occurs on a covalently modified surface (a bead or flat silicon surface) with complementary linkers covalently attached to it, using a specific dilution of library fragments as input. Panel B represents the stepwise sequencing process whereby reagents are introduced to extend the primed fragments, the incorporated fluorescent nucleotides are detected, the 3 end is deblocked, and the fluorescent groups on the incorporated nucleotides removed prior to the next stepwise sequencing-by-synthesis series. This fact underlies an important concept for digital sequencing methods: the number of specific sequencing reads generated is directly proportional to the amount of input nucleic acid, accurately reflecting amplified regions of a genome, for example. Solexa marketed the first commercially available sequencer using this technology in 2006, and was acquired by Illumina in 2007. The limitations on read length are primarily a signal-to-noise issue, where increasing numbers of steps in the sequencing-by-synthesis approach produces increasing noise at each step that competes with true signal detection. Hence, the data quality of Illumina reads tends to decrease with increasing step numbers. Paired end reads of this type physically are linked and defined by the fragment size, permitting their accurate placement onto the reference genome by alignment, and effectively permitting more reads to contribute to coverage from a given sequencer run (when compared to single-end reads). Upon incorporation, there is a release of hydrogen ions that are detected by the pH-sensing capability of the chip, detected in panel C. The flow of the four nucleotides occurs in a stepwise fashion, with a detection step and an intervening wash. This approach follows for all wells containing beads on the Ion Chip, resulting in massively parallel sequencing. As with the Illumina technology, read lengths are short, in the 100 to 400 bp range. Unlike the Illumina platform that uses paired-end reads, Ion Torrent sequencing reads are single-end reads. The source of most sequencing errors generated by the Ion Torrent platform are insertion/ deletion errors in stretches of identical bases on the template strand as a result of the difficulty of discerning the pH change ratio associated with incorporation of the same nucleotide above four consecutive identical nucleotides. The output, read length, run time, and cost vary by the Ion Chip type used (up to 2 Gb). The other major difference in the Pacific Biosciences approach is in the read length obtained, which ranges according to the template type but can exceed 50,000 bases with the input of very long molecules to the library construction. The sequencing process initiates with the introduction of fluorescent nucleotides and buffers, and is continuously monitored by the excitation/detection optics during the run time. As fluorescently tagged nucleotides sample into the active site, they can be detected with sufficient dwell time upon their incorporation into the synthesized strand. Because each fluorescent group is specific to the nucleotide identity, the sequence is read out based on the detected emission wavelength. Single-molecule sequencing has, by definition, an inherently higher error rate as a consequence of the signal-to-noise ratio associated with detecting a single event in real time. The predominant error type in Pacific Biosciences sequencing reads is an insertion/deletion error that may be a result of inaccuracies in detecting (1) a nucleotide that had a longer than average dwell time but was not incorporated, (2) a single nucleotide that incorporated but was mistaken for two (or more) nucleotides, or (3) by errors in detecting multiple nucleotide incorporations into a homopolymer stretch. Assembly has obvious advantages in its ability to represent novel content in a genome and to provide long-range haplotyping information. Nanopore sequencing, while still somewhat theoretical, may offer rapid sequencing with very long read lengths. As the throughput per run improved, larger genomes, including human genomes, were studied, including the first cancer genome. By combining a whole-genome library with the hybrid capture probes under conditions that favor hybridization (stoichiometry of probes to targets, temperature, and buffer conditions), probe:library fragment hybrids are formed. Following their selection by streptavidin magnetic bead binding and application of a magnetic force to isolate the beads, the noncaptured library fragments are removed, washes performed and the hybridized fragments eluted by denaturation from the probes. Although exome sequencing costs about one-tenth of whole-genome sequencing, it is important to note that typical yields from hybrid capture range from 85 to 90 percent of the targeted regions being covered at sufficient depth to confidently predict variants. In general, probes are designed for the targeted regions of interest, which can constitute a small number of genes or hotspot loci, up to the full exome (all annotated genes in a genome). Following hybrid capture, the probe:library duplexes are isolated from solution by streptavidin magnetic beads. Release of the library fragments by denaturation is followed by amplification, quantitation, and sequencing. Single nucleotide variants and short insertion/deletion variants can be detected, but copy number and structural variants are difficult to detect reliably, especially if they are not anticipated by the addition of specially designed probes to capture them and by the specialized analyses required to detect them. It also is a result of computational infrastructure and software pipeline requirements to align and analyze data because of the sheer magnitude of data generated in a single experiment, which is exacerbated by multiple samples, multiple time points, and the need to integrate data of different types for the correlative analyses that are desired. In practice, the desired comparison (whether the sequencing platform is a targeted gene panel, exome, or whole genome) is achieved by first aligning sequencing reads from the tumor library and from the matched normal library against the human genome reference sequence as separate entities. Algorithms that have specialized logic to identify different types of variation (single nucleotide, or "point" mutations, small insertions or deletions, copy number, or structural alterations) then are used to separately examine each set of read alignments and to identify the specific variation type relative to the human genome reference sequence. Lastly, the resulting variants that are identified are compared between the tumor and normal datasets, to identify those variants that appear unique to the tumor. As a means of interpreting the impact of all identified somatic variants on the sequence of amino acids in a given gene, for example, one must secondarily apply the annotation of the human genome onto identified single nucleotide and indel (term for the insertion or the deletion of bases) variants that occur within the coding regions and splice sites of known genes. Indel mutations typically cause a shift in the open reading frame ("frameshift") and result in a different amino acid sequence and length of the resulting protein, depending upon the number of added or deleted nucleotides. If the number added or deleted is a multiple of three nucleotides, the open reading frame is preserved but the protein sequence is altered accordingly.
Order serophene overnight
Desferrioxamine is a naturally occurring iron-chelating compound elaborated by the microorganism Streptomyces pilosus breast cancer triple negative serophene 50mg for sale, having evolved to enable the microbe to obtain iron from its environment. Urine iron is derived primarily from red cells broken down by macrophages, whereas fecal iron is believed to be from iron chelated in the liver. In the early Chapter 43: Iron Deficiency and Overload 645 20th century, the diagnosis was reserved for the rare patient with fullblown bronzed diabetes. Today, the diagnosis is applied to any person found to be homozygous for the C282Y mutation, or, indeed, anyone with an increased transferrin saturation and elevated serum ferritin level. In reality, patients with a diagnosis of hemochromatosis based on genetic and/or biochemical criteria have a normal life span. This is not to suggest that patients do not die of hereditary hemochromatosis; it is simply that the penetrance of the disorder as detected on genetic or biochemical bases is so low that the few deaths that do occur cannot be detected even in very sizable series. For those patients with classical hereditary hemochromatosis who are clinically affected, it is likely that removal of iron by phlebotomy prevents further complications and prolongs life span. Although controlled studies of the effect of phlebotomy are not ethically feasible, serial observations in patients undergoing phlebotomy suggest that cirrhosis is either stabilized or may, at least in some patients, improve. Cardiac deaths seem to be particularly common,276 and in a few cases cardiac transplantation has been performed successfully, but there are insufficient data concerning this rare disorder to allow one to provide more precise information about the outlook. Institution of iron chelation has greatly improved outcomes in -thalassemia major and similar disorders, but the prognosis is grim when iron chelation is not performed (Chap. Death is most frequently a result of cardiac failure but this complication is preventable with modern chelation regimens. Hershko C, Skikne B: Pathogenesis and management of iron deficiency anemia: Emerging role of celiac disease, Helicobacter pylori, and autoimmune gastritis. Stewart, Termanini, Sutliff, et al: Iron absorption in patients with Zollinger-Ellison syndrome treated with long-term gastric acid antisecretory therapy. Panzuto F, Di Giulio E, Capurso G, et al: Large hiatal hernia in patients with iron deficiency anaemia: A prospective study on prevalence and treatment. Haurani C, Carlin A, Hammoud Z, et al: Prevalence and resolution of anemia with paraesophageal hernia repair. Karamanou M, Androutsos G: Lasthenie de Ferjol syndrome: A rare disease with fascinating history. Hirayama Y, Sakamaki S, Tsuji Y, et al: Fatality caused by self-bloodletting in a patient with factitious anemia. Domellof M, Braegger C, Campoy C, et al: Iron requirements of infants and toddlers. Schmitz U, Ko Y, Seewald S, et al: Iron-deficiency anemia as the sole manifestation of celiac disease. Pagani A, Nai A, Corna G, et al: Low hepcidin accounts for the proinflammatory status associated with iron deficiency. Sachdev H, Gera T, Nestel P: Effect of iron supplementation on physical growth in children: Systematic review of randomised controlled trials. Wang B, Zhan S, Gong T, et al: Iron therapy for improving psychomotor development and cognitive function in children under the age of three with iron deficiency anaemia. Abdullah K, Kendzerska T, Shah P, et al: Efficacy of oral iron therapy in improving the developmental outcome of pre-school children with non-anaemic iron deficiency: A systematic review. Hermoso M, Vucic V, Vollhardt C, et al: the effect of iron on cognitive development and function in infants, children and adolescents: A systematic review. Hornyak M, Scholz H, Kohnen R, et al: What treatment works best for restless legs syndrome Cortese S, Angriman M, Lecendreux M, et al: Iron and attention deficit/hyperactivity disorder: What is the empirical evidence so far Deloche C, Bastien P, Chadoutaud S, et al: Low iron stores: A risk factor for excessive hair loss in non-menopausal women. Verma V, Ayalew G, Sidhu G, et al: An analysis of the relationship between severe iron deficiency anemia and thrombocytopenia. Garby L, Irnell L, Werner I: Iron deficiency in women of fertile age in a Swedish community. Efficiency of several laboratory tests to predict the response to iron supplementation. Geisser P, Burckhardt S: the pharmacokinetics and pharmacodynamics of iron preparations. Medrano-Engay B, Irun P, Gervas-Arruga J, et al: Iron homeostasis and infIammatory biomarker analysis in patients with type 1 Gaucher disease. Mekinian A, Stirnemann J, Belmatoug N, et al: Ferritinemia during type 1 Gaucher disease: Mechanisms and progression under treatment. Moore C Jr, Ormseth M, Fuchs H: Causes and significance of markedly elevated serum ferritin levels in an academic medical center. Uaprasert N, Rojnuckarin P, Bhokaisawan N, et al: Elevated serum transferrin receptor levels in common types of thalassemia heterozygotes in Southeast Asia: A correlation with genotypes and red cell indices. Suominen P, Punnonen K, Rajamaki A, et al: Serum transferrin receptor and transferrin receptor-ferritin index identify healthy subjects with subclinical iron deficits. Punnonen K, Irjala K, Rajamaki A: Serum transferrin receptor and its ratio to serum ferritin in the diagnosis of iron deficiency. Brugnara C, Mohandas N: Red cell indices in classification and treatment of anemias: From M. Brugnara C, Schiller B, Moran J: Reticulocyte hemoglobin equivalent (Ret He) and assessment of iron-deficient states. Beutler E, West C: Hematologic differences between African-Americans and whites: the roles of iron deficiency and alpha-thalassemia on hemoglobin levels and mean corpuscular volume. Duma H, Efremov G, Sadikario A, et al: Study of nine families with haemoglobinLepore. Cartei G, Chisesi T, Cazzavillan M, et al: Relationship between Hb and HbA2 concentrations in beta-thalassemia trait and effect of iron deficiency anaemia. Infusino I, Braga F, Dolci A, et al: Soluble transferrin receptor (sTfR) and sTfR/log ferritin index for the diagnosis of iron-deficiency anemia. Intragumtornchai T, Rojnukkarin P, Swasdikul D, et al: the role of serum ferritin in the diagnosis of iron deficiency anaemia in patients with liver cirrhosis. Prieto J, Barry M, Sherlock S: Serum ferritin in patients with iron overload and with acute and chronic liver diseases. Aslan D, Crain K, Beutler E: A new case of human atransferrinemia with a previously undescribed mutation in the transferrin gene. Chen C, Wen S, Tan X: Molecular analysis of a novel case of congenital atransferrinemia. Lucotte G, Dieterlen F: A European allele map of the C282Y mutation of hemochromatosis: Celtic versus Viking origin of the mutation Tsukamoto H, Horne W, Kamimura S, et al: Experimental liver cirrhosis induced by alcohol and iron. Arosio P, Ingrassia R, Cavadini P: Ferritins: A family of molecules for iron storage, antioxidation and more. Tamary H, Shalev H, Perez-Avraham G, et al: Elevated growth differentiation factor 15 expression in patients with congenital dyserythropoietic anemia type I. Lanzara C, Roetto A, Daraio F, et al: Spectrum of hemojuvelin gene mutations in 1q-linked juvenile hemochromatosis. Vaiopoulos G, Papanikolaou G, Politou M, et al: Arthropathy in juvenile hemochromatosis. Ko C, Siddaiah N, Berger J, et al: Prevalence of hepatic iron overload and association with hepatocellular cancer in end-stage liver disease: Results from the National Hemochromatosis Transplant Registry. Ogimoto M, Anzai K, Takenoshita H, et al: Criteria for early identification of aceruloplasminemia. In contrast, characteristics of anemia that may occur with deficiencies of the other vitamins and minerals are poorly defined and relatively rare in humans. When present, they usually exist not as isolated deficiencies of one vitamin or one mineral, but rather, as a combination of deficiencies resulting from malnutrition or malabsorption. In this context, it is difficult to deduce which abnormalities are the result of which deficiency.
Serophene 50 mg discount
These are frequently updated in software or when new models are introduced to improve sensitivity and specificity womens health diet order serophene 50mg otc. In this way, instruments identify samples that contain cells or abnormalities the instrument cannot definitively identify, so that a skilled morphologist can visually evaluate that specimen. Some of these flags can be adjusted or suppressed by the user to achieve an appropriate balance that minimizes both false positives and false negatives. Guidelines for manual smear review based on comparative data have been published, based on instruments then in common use. Depending on workload and space considerations, laboratories may choose to link automated hematology analyzers with automated blood film preparation and automated image analyzers to facilitate manual morphologic review of cells by traditional light microscopy or online review of digitized images. Quantitative measures available from automated cell counters are reliable and provide a rapid and cost-effective way to screen for primary or secondary disturbances of hematopoiesis. Light microscopic observation of the blood film is essential to confirm certain quantitative results and to investigate qualitatively abnormal differentiation of the hematopoietic lineages. Based on examination of the blood, the physician is directed toward a more focused assessment of marrow function or to systemic disorders that secondarily involve the hematopoietic system. Over the past several decades, instruments have become increasingly sophisticated with the use of multiple parameters to produce more precise results in the great majority of patient samples. In a typical automated hematology analyzer, the blood sample is aspirated and separated into different fluidic streams. The streams are mixed with various buffers that accomplish specific purposes in the analysis, for instance, using differential lysis to distinguish subsets of leukocytes, reagents to measure hemoglobin or detect myeloperoxidase containing leukocytes, and various fluorescent dyes. Measurements of each fluidic stream are made in flow as the sample passes through a series of detectors in what are essentially modified flow cytometers (Chap. Commonly used principles include light scatter at various angles, electrical impedance and conductivity, and fluorescence or light absorption of cells stained in flow. Light scatter yields information about cell size (using scatter at low-incident angles), nuclear lobulation, and cytoplasmic granularity (using high-angle light scatter) and refractive index, with polarization of the scattered light as an additional parameter. If red cells are converted to spherocytes by the buffer solution to eliminate the variability of cell shape, light scatter at different angles can provide information about hemoglobin content, as well as size of individual red cells. Cell size is also estimated by measuring change in electrical resistance, which is proportional to cell size as cells enter a narrow orifice through which a direct current is maintained, the original Coulter principle, named for Wallace Coulter who developed the electronic particle counter. Differential lysis with detergents of varying strength or pH is used to separate certain leukocyte types, such as basophils and immature granulocytic cells, from the major normal blood cell types. Light absorption is the principle used for hemoglobin measurement and in some instruments for identifying peroxidase-positive granulocytes. There is significant overlap in methodology between automated hematology analyzers and flow cytometers (flow cytometers are discussed in Chap. The latter are distinguished by extensive use of fluorochrome tagged antibodies to identify cell subtypes. These instruments have replaced laborious manual work, but also demand increasing interpretation skills on the part of laboratory technologists. Automated blood analyzers have been adapted to accurately count the smaller numbers of blood cells typically found in body fluids,12 but accurate differential counts13 and detection of blast cells in fluids of patients14 remains a challenge. Point of care "bedside" testing is far more challenging in hematology than for typical clinical chemistry analytes for many of the reasons described above. More work remains to be done to demonstrate the reliability and clinical impact of such testing strategies. Schematic of multiparameter cell discrimination in an automated hematology analyzer. Examples of how samples containing various Basophils A abnormal findings are flagged for manual review. Multiple flags, including cells in the area of atypical lymphocytes, and platelet clumps with abnormal platelet volume distribution. This figure is not intended as a comprehensive illustration of the technical details, but serves to demonstrate that differential lysing reactions coupled with multiparameter lightscatter, impedance, capacitance, and fluorescence measurements are used to analyze blood cells in current high-throughput instruments. Measurement of the Red Cell Count and Hematocrit the hematocrit may also be determined by subjecting the blood to sufficient centrifugal force to pack the cells while minimizing trapped extracellular fluid. This approach was traditionally done in capillary tubes filled with blood and centrifuged at very high speed in a small tabletop centrifuge, and the technique was referred to as the "microhematocrit" or informally as a "spun crit. However, this is a manual procedure not well adapted to routine processing in a high-volume clinical laboratory, and is affected by varying amounts of plasma trapped between red cells in the packed cell volume,16 typically about 2 to 3 percent of the packed volume. Hemoglobin is intensely colored, and this property has been used in methods for estimating its concentration in blood. Erythrocytes contain a mixture of hemoglobin, oxyhemoglobin, carboxyhemoglobin, methemoglobin, and minor amounts of other forms of hemoglobin. To determine hemoglobin concentration in the blood, red cells are lysed and hemoglobin variants are converted to the stable compound cyanmethemoglobin for quantification by absorption at 540 nm. All forms of hemoglobin are readily converted to cyanmethemoglobin except sulfhemoglobin, which is rarely present in significant amounts. In automated blood cell counters, hemoglobin is usually measured by a modified cyanmethemoglobin or an alternate lauryl sulphate method. In practice, the major interference with this measurement is chylomicronemia, but newer instruments identify and minimize this interference. Noninvasive transcutaneous monitoring of total hemoglobin concentration, as well as methemoglobin and carboxyhemoglobin, using multiwavelength pulse oximetry has become available. After the first week or two of extrauterine life, the hemoglobin falls from levels of approximately 17 g/dL to levels of approximately 12 g/dL by 2 months of age. Thereafter, the levels remain relatively constant throughout the first year of life. The size and hemoglobin content of erythrocytes (red cell indices), based on population averages, have traditionally been used to assist in the differential diagnosis of anemia. These red cell indices are average quantities and, therefore, may not detect abnormalities in blood with mixed-cell populations. The number of reticulocytes in a volume of blood permits an estimate of marrow erythrocyte production and is thus useful in evaluating the pathogenesis of anemia by distinguishing inadequate production from accelerated destruction (Chap. The manual method for enumerating reticulocytes by placing a sample of blood in a tube containing new methylene blue and preparing a blood film to enumerate the proportion of cells that show blue beaded precipitates (residual ribosomes) has largely been replaced by automated methods, which are incorporated into high-volume hematology analyzers. Various proprietary combinations of light scatter and other parameters are used to minimize interferences such as nucleated red cells, nuclear remnants (Howell-Jolly bodies), malaria parasites, or platelet clumps. Automated reticulocyte counts are typically reported in absolute numbers (reticulocytes per L or per L of blood), obviating the need to correct for a reduced red cell count (anemia), if present. Normal ranges should be validated by the clinical laboratory for the specific methods in use. The correlation between manual and automated methods of reticulocyte enumeration is good, but reference ranges differ slightly among the methods, given the different dyes and conditions used and the continuous nature of the variables separating reticulocytes from mature red cells. A limitation at present is that the methods lack standardization and reference ranges for these parameters are instrument dependent. The result is a plethora of new indices that are in many cases specific to an instrument manufacturer, presenting new diagnostic opportunities but also a confusing nomenclature and a potential lack of comparability. New formulas for distinguishing causes of microcytosis based on several novel red cell indices function about as well45 or somewhat better46 than traditional formulas for differentiating iron deficiency from thalassemia trait. More sophisticated mathematical modeling of individual cell-based volume and hemoglobin content data available in current analyzers has been used in a systems biology approach to demonstrate latent iron deficiency and to distinguish causes of microcytosis. The theoretical advantage is that acute changes in red cell function would be detected more rapidly and reliably in the reticulocyte fraction as opposed to the total red cell population. These parameters have the advantage of ready access in the context of an automated blood count, but the availability of differently derived and calculated parameters from various instrument makers is a challenge to remember and compare across laboratories. Nucleated Red Cells Nucleated red cells are present in newborns, particularly if physiologically stressed, and in a variety of disorders, including hypoxic states (congestive heart failure), severe hemolytic anemia, primary myelofibrosis, and infiltrative disease of the marrow (Chap. Most modern automated hematology analyzers are capable of detecting and quantitating nucleated red blood cells, which were a source of spuriously elevated leukocyte counts in earlier instruments, at a level of 1 to 2 nucleated red cells per 100 leukocytes. Malarial Parasites Malarial parasites can also be detected by some current analyzers, based on detecting parasite infected red cells or neutrophils containing ingested hemozoin in regions of the multiparameter display that are not characteristically populated in normal blood (sometimes causing spurious eosinophilia57). Some reports indicate high sensitivity and specificity with certain instrumentation,58 a useful consideration in endemic areas where access to technologists with morphologic expertise may not be consistent. Other Abnormalities Not Detected by Automation Some disorders, such as immune and hereditary spherocytosis (Chaps. Manual counting of leukocytes is used only when the instrument reports a potential interference or the count is beyond instrument linearity limits.
Buy cheap serophene 25 mg
Because 1 mL of packed red cells contains approximately 1 mg of iron women's health clinic fort qu'appelle cheap 100mg serophene with visa, a daily plasma iron turnover of 0. However, an elevated serum iron concentration gives erroneous impressions of the state of erythropoiesis. Moreover, more prolonged sampling of plasma following an intravenous injection of 59 Fe has shown that clearance is not a single exponential, but must be represented by several exponential components. Careful analysis of such models has generated computer-supported methods calculating the degree and effectiveness of erythroid activity. Moreover, even these sophisticated methods may not give an accurate account of the state of erythropoiesis. Despite a constant rate of red cell production, the plasma iron turnover was found to increase with increasing plasma iron and transferrin saturation. This finding was first thought to result from increased nonerythroid iron uptake and led to the introduction of various correction factors in the calculation of red cell iron turnover. Consequently, total plasma iron turnover depends on the degree of saturation and does not necessarily reflect the number of transferrin receptors, presumably a critical measure of erythropoietic capacity. Garofalo F, Pellegrino D, Amelio D, et al: the antarctic hemoglobinless icefish, fifty-five years later: A unique cardiocirculatory interplay of disaptation and phenotypic plasticity. Palis J, Robertson S, Kennedy M, et al: Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Yoshida H, Kawane K, Koike M, et al: Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Soni S, Bala S, Gwynn B, et al: Absence of erythroblast macrophage protein (emp) leads to failure of erythroblast nuclear extrusion. Evans T, Felsenfeld G: the erythroid-specific transcription factor eryf1: A new finger protein. Gregory T, Yu C, Ma A, et al: Gata-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xl expression. Nuez B, Michalovich D, Bygrave A, et al: Defective haematopoiesis in fetal liver resulting from inactivation of the eklf gene. Robb L, Lyons I, Li R, et al: Absence of yolk sac hematopoiesis from mice with a targeted disruption of the scl gene. Uda M, Galanello R, Sanna S, et al: Genome-wide association study shows bcl11a associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Angelillo-Scherrer A, Burnier L, Lambrechts D, et al: Role of gas6 in erythropoiesis and anemia in mice. Parganas E, Wang D, Stravopodis D, et al: Jak2 is essential for signaling through a variety of cytokine receptors. Walrafen P, Verdier F, Kadri Z, et al: Both proteasomes and lysosomes degrade the activated erythropoietin receptor. Nakamura Y, Nakauchi H: A truncated erythropoietin receptor and cell death: A reanalysis. Beck I, Ramirez S, Weinmann R, et al: Enhancer element at the 3-flanking region controls transcriptional response to hypoxia in the human erythropoietin gene. Cavill I, Trevett D, Fisher J, et al: the measurement of the total volume of red cells in man: A non-radioactive approach using biotin. Recommended methods for measurement of red-cell and plasma volume: International committee for standardization in haematology. Bauer W, Stray S, Huebers H, et al: the relationship between plasma iron and plasma iron turnover in the rat. Beguin Y: the soluble transferrin receptor: Biological aspects and clinical usefulness as quantitative measure of erythropoiesis. Such studies show that normal human red cells have a finite life span averaging 120 days, with very little random destruction. The mitochondrial and ribosomal removal highlighting maturation of the reticulocyte is accompanied by increasing cell density, but after a few days of intravascular life span there is little further increase in density or other changes in the physical property of the red cells. This has made the senescent changes in the red cell that mark it for destruction difficult to study. Candidates for such changes include changes in membrane band 3 and exposure of phosphatidylserine on the membrane, which may be of major importance. In 1946, Shemin and Rittenberg demonstrated that the incorporation of nitrogen (15N)-labeled glycine into heme could be used to measure the life span of the red cells. These can be divided into three groups: (1) those that label a cohort of cells, (2) those that label cells randomly, and (3) those that use indirect measurements such as the rate of production of red cells or the rate of heme breakdown. The first two methodologic approaches yield information about the nature of the shortening of the red cell life span, age-dependent or random. The labels used are glycinecontaining labeled 15N,14 radioactive carbon (14C),15 or radioactive iron (either 55Fe or 59Fe). In addition, radioiron from destroyed red cells may be reused, making it difficult to interpret results. Furthermore, the increasing restrictions on use of radiochemicals has drastically decreased availability of these two previously widely used nuclear medicine tests. A simple double-labeling technique that allows nonradioactive cohort labeling was described using two distinct labeling steps separated by a defined time interval. The initial labeling step uses biotin that binds to all circulating cells (the red blood cells accounting for most of the label) the second administered labeling substance at later time digoxigenin then distinguishes erythrocyte subpopulation of known age. This is an extremely efficient process as macrophages phagocytose approximately 5 million erythrocytes every second without a significant release of hemoglobin into the circulation. The precise molecular mechanism by which macrophages recognize senescent red blood cells for phagocytosis remains largely unknown. As red blood cells age, several physiologic changes occur that may serve as signals for recognition by macrophages. Chromium-51 Method By far the most commonly used radioactive isotope for the measurement of the red cell life span is 51Cr. As the chromate ion penetrates the red cell membrane it binds to the and chains of globin. If the data indicate exponential disappearance and it is necessary to use a semilogarithmic paper in order to depict the data on a straight line, the destruction is random and the life span is 1. One objection to this method is that the degree of chromium elution is not a constant but varies from day to day and is influenced by various disease states. Although computerassisted methods can resolve ambiguities, the inherent biologic and technical variations in measuring red cell life span are such that it is better to rely on chromium T1/2 with intuitive adjustments based on clinical findings. In addition to being a nonradioactive probe, biotin labeling has other advantages. The transfused cells can be isolated from the patients on avidin substrates for further characterization. Biotin labeling has been used to demonstrate that sickle cells without fetal hemoglobin have a shorter in vivo survival compared to those with fetal hemoglobin,37 and has been also instrumental in showing a role for phosphatidylserine exposure in the clearance of sickle cells. When red cells are labeled randomly with chromium-51 (51Cr) there is a daily 1 percent elution that needs to be corrected for in the calculation of total red cell life span. The life span is estimated by measuring the survival of randomly labeled red cells. This takes normally approximately 5 minutes,32 but may be longer in patients with splenomegaly. Following equilibration, the cells that have been damaged by the labeling process will be removed from the circulation during the next 24 hours. Fortunately, it is usually possible to gain an accurate estimate of red cell half-life by sampling three times a week for 1 to 2 weeks. In the normal human the red cell, life span is finite with an average of approximately 120 days, with very little random destruction, that is, loss irrespective of cell age (0. For clinical use, the red cell life span is usually expressed as chromium half-life (T1/2) and compared to the normal value for the method of 30 days.
Diseases
- Chromosome 14, trisomy mosaic
- Pyridoxine deficit
- Moerman Van den berghe Fryns syndrome
- Oral-pharyngeal disorders
- Buttiens Fryns syndrome
- Neuroaxonal dystrophy, late infantile
- Testes neoplasm
- Loiasis
- Vitamn B12 responsive methylmalonicaciduria
25mg serophene with visa
Finally womens health diet pill order genuine serophene online, and most importantly, absolute polycythemia is not normovolemic but is accompanied by increased blood volume, which, in turn, enlarges the vascular bed and decreases peripheral resistance. Because blood pressure remains stable, the increased blood volume must be associated with increased cardiac output and increased oxygen transport (cardiac output times hemoglobin concentration). These curves show that hypervolemia per se increases oxygen transport and that the optimum oxygen transport in these conditions occurs at higher hematocrit values than in normovolemic states. Consequently, despite the increased viscosity, a moderate increase in hematocrit is beneficial. Uncorrected Tissue Hypoxia A certain residual degree of tissue hypoxia remains despite mobilization of compensatory mechanisms. Hypoxia is essential for initiation of adequate cardiovascular and erythropoietic compensation mechanisms, but severe tissue hypoxia can cause the following symptoms: dyspnea on exertion or even at rest; angina; intermittent claudication; muscle cramps, typically at night; headache; light-headedness; and fatigue. Relative anemia is characterized by a normal total red cell mass in an increased plasma volume, resulting in a dilution anemia, a disturbance in plasma volume regulation. However, dilution anemia is of clinical and differential diagnostic importance for the hematologist. Classification of the absolute anemias with decreased red cell mass is difficult because the classification has to consider kinetic, morphologic, and pathophysiologic interacting criteria. Anemia of acute hemorrhage is not a diagnostic problem and is usually a genitourinary or gastrointestinal matter, not a hematologic consideration. Initially, all anemias should be divided into anemias caused by decreased production and anemias caused by increased destruction of red cells. Subsequent diagnostic breakdown can be based on either morphologic or pathophysiologic criteria. Morphologic classification subdivides anemia into (1) macrocytic anemia, (2) normocytic anemia, and (3) microcytic hypochromic anemia. In addition, anemia resulting from vitamin or iron-deficiency states occurs in a significant proportion of patients with normal red cell indices. This chapter presents a classification based on our present concepts of normal red cell production and red cell destruction. Therapeutic intervention depends on identifying the defective step and instituting the specific therapy. Under normal conditions, the rate of red cell production is adjusted to maintain the red cell mass at approximately 30 mL per kilogram of body weight. Because the life span of red cells in polycythemia is normal, a doubling of the daily rate of red cell production is adequate to maintain a polycythemic red cell mass of 60 mL/kg. Pluripotential hematopoietic stem cell failure (1) Autoimmune (Aplastic anemia) (Chap. Erythroid progenitor cell failure (1) Pure red cell aplasia (parvovirus B19 infection, drugs, associated with thymoma, autoantibodies, etc. Functional impairment of erythroid and other progenitors from nutritional and other causes (1) Megaloblastic anemias (Chap. Mechanical (1) Macroangiopathic (march hemoglobinuria, artificial heart valves [Chap. Chemical injury and complex chemicals (arsenic, copper, chlorate, spider, scorpion, and snake venoms, etc. Red cell enzyme defects (pyruvate kinase, 5 nucleotidase, glucose-6-phosphate dehydrogenase deficiencies, other red cell enzyme disorders [Chap. Porphyrias (congenital erythropoietic and hepatoerythropoietic porphyrias, rarely congenital erythropoietic protoporphyria [Chap. In erythrocytosis, the number of red cells destroyed daily merely causes a slight increase in bilirubin levels. The presence of secondary gout and splenomegaly are usually signs of a myeloproliferative neoplasm rather than of erythrocytosis alone. The increased viscosity and expansion of vascular space are responsible for many of the signs and symptoms of polycythemia. The characteristic rubor in patients with polycythemia vera is caused by excessive deoxygenation of blood flowing sluggishly through dilated cutaneous vessels. Nonspecific symptoms such as headaches, dizziness, tinnitus, and a reported feeling of fullness of the face and head probably are caused by a combination of increased viscosity and vascular dilatation. Outline of the process of differentiation, proliferation, and maturation underlying the production and destruction of red blood cells. In the presence of adequate amounts of nutrients, such as vitamin B12, folic acid, and iron, precursor cells proliferate and mature into nucleated red cells, reticulocytes, and mature red blood cells. Oxygen transport is computed from Hct and O2 flow (1/viscosity) and is recorded in arbitrary units. Hypoxemia (1) Chronic lung disease (2) Sleep apnea (3) Right-to-left cardiac shunts (4) High altitude (5) Smoking b. High erythropoietin polycythemias caused by mutations of von Hippel-Lindau gene other than Chuvash mutation c. Thrombosis are common in polycythemia vera, but are not seen at similar frequencies in other types of polycythemias (Chaps. Coronary blood flow is decreased in polycythemia,34 so the risk of coronary thrombosis in patients with a high hematocrit is assumed to be increased; however, statistical analyses have yielded equivocal evidence of such a relationship. In polycythemia vera, however, it has been advocated that normalization of red cell mass should be accomplished before surgery; again, firm data supporting this practice are lacking (Chap. Differentiation of absolute from relative polycythemia can be difficult at hematocrits of less than 60 percent. Bohr C, Hasselbalch K, Krogh A: [Concerning a biologically important relationship: the influence of the carbon dioxide content of blood on its oxygen binding]. Oxygen transport at various hematocrit levels in nor- movolemia, mild hypervolemia, and severe hypervolemia. The circulatory effects of hematocrit variations in normovolemic and hypervolemic dogs. All reported polycythemic heterozygotes for Chapter 34: Clinical Manifestations and Classification of Erythrocyte Disorders 511 32. Neutropenia, monocytopenia, and thrombocytopenia, when severe, are life-threatening because of the risk of infection and bleeding, complicated by severe anemia. The disorder also can occur after (1) prolonged high-dose exposure to certain toxic chemicals. The final common pathway may be through cytotoxic T-cell autoreactivity, whether idiopathic or associated with an inciting agent since they all respond in a similar fashion to immunosuppressive therapy. The differential diagnosis of acquired aplastic anemia includes a hypoplastic marrow that can accompany paroxysmal nocturnal hemoglobinuria or hypoplastic oligoblastic (myelodysplastic syndrome) or polyblastic myelogenous leukemia. The disease may be significantly ameliorated or occasionally cured by immunotherapy, especially a regimen coupling antithymocyte globulin with cyclosporine. However, after successful treatment with immunosuppressive agents, the disease may relapse or evolve into a clonal myeloid disorder, such as paroxysmal nocturnal hemoglobinuria, a clonal cytopenia, or oligoblastic or polyblastic myelogenous leukemia. Several uncommon inherited disorders, including Fanconi anemia, Shwachman-Diamond syndrome, dyskeratosis congenita and others have as a primary manifestation aplastic hematopoiesis. The decrease in hematopoiesis results in reticulocytopenia, anemia, granulocytopenia, monocytopenia, and thrombocytopenia. The diagnosis usually requires the presence of pancytopenia with a neutrophil count fewer than 1500/L (1. Most cases of aplastic anemia are acquired; fewer cases are the result of an inherited disorder, such as Fanconi anemia, Shwachman-Diamond syndrome, and others (see "Hereditary Aplastic Anemia" below). Thrombocytopenia was difficult to measure and the role of blood dust (platelets) was controversial at that time.
Order serophene 25mg on line
Alternatively women's health clinic spruce grove order discount serophene on line, iron may be released from ferritin by autophagy followed by lysosomal degradation. Microscopically, in unstained tissue sections or marrow films it appears as clumps or granules of golden refractile pigment. Under pathologic conditions, it may accumulate in large quantities in almost every tissue of the body. Hemosiderin is chemically similar to the iron core of ferritin and may be derived from ferritins whose protein shells have been digested in lysosomes. In women, iron absorbed must be sufficient to replace that lost through menstruation or diverted to the fetus or milk during and after pregnancy. Oxalates, phytates, and phosphates complex with iron and retard its absorption, whereas simple reducing substances, such as hydroquinone, ascorbate, lactate, pyruvate, succinate, fructose, cysteine, and sorbitol, increase iron absorption. It is present in small amounts in all skeletal and cardiac muscle cells, where it may serve as an oxygen reservoir to protect against cellular injury during periods of oxygen deprivation and may scavenge nitric oxide and reactive oxygen species. Some of the iron reenters plasma, causing a biphasic curve of 59 Fe clearance 1 to 2 days after injection. The change in slope defines the size of the labile pool, normally 80 to 90 mg of iron. Although a small compartment, it is an extremely vital one and is sensitive to iron deficiency. Chapter 42: Iron Metabolism 619 the transit time, and mucus secretion all play roles in iron absorption. Red wine, contrary to popular belief, inhibits iron absorption, probably because of the presence of polyphenols. The amount of iron absorbed is normally tightly regulated according to body needs. Active erythropoiesis and/or iron deficiency increase absorption; iron overload and systemic inflammation decrease absorption. Accidental or deliberate ingestion of large doses of medicinal iron can therefore cause iron intoxication. Iron from other cell types is likely also recycled, but this source contributes little to iron flux and has not been studied. Destruction of aged erythrocytes and hemoglobin degradation occur within macrophages (Chap. This proceeds at a rate sufficient to release approximately 20 percent of the hemoglobin iron from the cell to the plasma compartment within a few hours. Thus, 20 to 70 percent of the hemoglobin iron of nonviable erythrocytes reappears in circulating red cells in 12 days. The remainder of the iron enters the storage pool as ferritin or hemosiderin and then turns over very slowly. In normal subjects, approximately 40 percent of this iron remains in storage after 140 days. When there is an increased iron demand for hemoglobin synthesis, however, storage iron may be mobilized more rapidly. Macrophages also take up the products of intravascular hemolysis, including hemoglobin (bound by haptoglobin) and heme (bound by hemopexin), using specific endocytic receptors for the complexes. Heme oxygenase 1 is mostly located in the endoplasmic reticulum in erythrophagocytic macrophages37 with the catalytic face in the cytosol, and little, if any, heme oxygenase in the phagosomal membrane. How much heme (if any) is exported intact by enterocytes and bound by plasma heme-binding protein hemopexin is not clear, but hemopexin knockout mice show minor retention of iron in duodenal enterocytes without any effect on systemic iron homeostasis,21 arguing against a major contribution from this mechanism, at least in mice. Efforts to identify the apical heme import mechanism in enterocytes have not yet been definitive. When the logarithm of the dose is plotted against the logarithm of the amount of iron absorbed, a rectilinear relationship is observed. Thus, at all levels, the greater the dose of iron, the more is absorbed, although the percent of the dose that is absorbed progressively declines. Schematic of iron uptake from the intestine and transfer to the plasma by an intestinal villus cell. Fe3+ is reduced to Fe2+ by ascorbic acid Ferroportin and apical membrane ferrireductases that include duodenal cytochrome b (dcytb). Basolateral export of Fe2 may be mediated + K by ferroportin in association with hephaestin. It has now become clear that intestinal iron absorption, plasma iron concentrations, and tissue distribution of iron are subject to endocrine regulation similar to that of other simple nutrients, for example, glucose or calcium, albeit in a somewhat more complex fashion. Hepcidin regulates plasma iron concentrations by controlling the absorption of iron by the intestinal epithelial enterocytes and its release from iron-recycling macrophages and hepatocytes involved in iron storage. The structural similarity of hepcidin and a class of antimicrobial peptides termed defensins suggests that the hormone evolved from the latter to modulate iron homeostasis as a mechanism of body defense against microorganisms. Overexpression of hepcidin results in marked iron-deficiency anemia in mice48 and a refractory anemia resembling the anemia of chronic inflammation in humans,49 and injection of synthetic hepcidin rapidly lowers plasma iron concentrations. In fact, patients with iron overload and high plasma iron levels are susceptible to such infections, such as with Yersinia enterocolitica (Chap. Hepcidin exerts its iron-regulatory effect by binding to ferroportin, a transmembrane iron-export protein expressed on enterocytes, macrophages, and hepatocytes. Once hepcidin has bound to ferroportin, the ferroportin is internalized and undergoes proteolysis. This results in decreased iron absorption from the gastrointestinal tract and a fall in the plasma iron concentration. For reasons that are not understood, involving perhaps the complex interactions of hepatocytes with other liver cells, isolated hepatocytes do not show consistently increased hepcidin synthesis after iron treatment, although small effects were observed when the cells were freshly harvested from mice. Fer- roportin is the only known transporter that exports iron from cells to plasma (and extracellular fluid). Hepcidin induces ferroportin endocytosis and proteolysis and thereby controls the transfer of iron to plasma from all its major sources: iron-absorbing duodenal enterocytes, iron-storing hepatocytes, and iron-recycling macrophages. Tmprss6 (also called matriptase 2) is a membrane serine protease that inhibits hepcidin transcription, likely by proteolysis of hemojuvelin. Later studies in mouse models56 provided evidence that the erythroid regulator is a marrow-derived suppressor of hepcidin. Erythroferrone is an erythropoietin-induced erythroblast-secreted glycoprotein that acts on hepatocytes to suppress their hepcidin production and is required for rapid suppression of hepcidin after hemorrhage or erythropoietin administration. Here it is removed from heme by heme oxygenase and released back into the plasma to repeat the cycle. The major function of the transport protein transferrin is to move iron from wherever it enters the plasma (intestinal villi, splenic and hepatic sinusoids) to the erythroblasts of the marrow and to other sites of use. Within hours after the onset of systemic infection, plasma iron concentration decreases. The response is thought to contribute to host defense, particularly against microbes with high dependence on environmental iron. Hypoferremia of inflammation is mediated by cytokine-induced increase in plasma hepcidin concentrations54 causing hepcidin-induced sequestration of iron in macrophages. Within the cytosol the holotransferrin-TfR1 complex is in a clathrin-coated vesicle. The vesicles fuse with endosomes and become acidified to pH 5 which releases iron from transferrin. Iron is tightly conserved in a nearly closed system in which each iron atom cycles repeatedly from plasma and extracellular fluid ("plasma") to the marrow, where it is incorporated into hemoglobin.
Cheap serophene 50mg
The lipid bilayer separates the erythrocyte cytoplasm from the external plasma environment and contains phospholipids and cholesterol menstrual cycle 7 days early order 25 mg serophene with amex, as well as integral transmembrane proteins, which are tethered to the skeleton by interactions with linker proteins. The choline phospholipids, phosphatidylcholine and sphingomyelin, are predominantly located in the outer leaflet and play a role in plasma lipid exchange and renewal of membrane phospholipids. Glycolipids carry several important red cell antigens, including A, B, H, and P, and are only found in the external leaflet with their carbohydrate moieties extending into the plasma. The aminophospholipids, phosphatidylserine and phosphatidylethanolamine, as well as phosphatidylinositol are located in the inner leaflet of the lipid bilayer. This asymmetric distribution of phospholipids is maintained by a dynamic process involving flippase and floppase enzymes, which translocate the aminophospholipids to the inner and outer leaflets, respectively. It activates the coagulation cascade and may contribute to thromboses4; it facilitates adhesion to the vascular endothelium; it provides a recognition signal for macrophages to phagocytose these cells; and it decreases the interaction of skeletal proteins with the bilayer, which destabilizes the membrane. Analysis of the individual proteins led to the renaming of some of them, such as band 1 and 2, which are now known as - and -spectrin, respectively. Technologic advances have enabled an in-depth analysis of the erythrocyte proteome by mass spectrometry, revealing a total of 340 membrane proteins. Integral or transmembrane proteins are embedded in the lipid bilayer by hydrophobic interactions and require detergents to extract them. They often protrude from the bilayer and extend into the plasma and/or the interior of the erythrocyte and these structural features correlate with their functions as transport proteins, receptors, signaling molecules, and carriers of red cell antigens. Peripheral proteins constitute the membrane skeleton and are loosely attached to the cytoplasmic face of the lipid bilayer and can be extracted by high or low salt concentrations or by high pH. Attachment is mediated indirectly by covalent or noncovalent interactions with the cytoplasmic domains of the transmembrane proteins, as well as by direct interactions with the inner leaflet of the lipid bilayer. These associations are dynamic and the affinity of binding is regulated by post-translational modifications of the proteins, including phosphorylation, methylation, glycosylation, or lipid modification (myristoylation, palmitoylation, or farnesylation). Peripheral proteins typically function either as structural proteins and form part of the membrane skeleton or they serve as linker proteins attaching the skeleton to the bilayer. Many erythrocyte proteins belong to superfamilies and have homologues in nonerythroid cells that are structurally related but are encoded by different genes. This genetic diversity explains why the clinical expression of most (but not all) red cell membrane protein mutations is confined to the erythroid lineage. Several proteins exist in different isoforms, created by tissue- and developmental stage-specific alternative splicing or by the use of alternative initiation codons or promoters. Many of the membrane proteins are large, multifunctional proteins and therefore the position of a mutation determines the functional abnormality and clinical phenotype. The anion exchange domain encompasses 13 helical transmembrane segments and one nonhelical segment all connected by hydrophilic loops. Phosphorylation at tyrosine 8 prevents binding, which liberates the active enzymes. This domain also serves as the major attachment Integral Membrane Proteins and C terminal regions of the protein extend into the cytoplasm and provide binding sites for several red cell proteins and enzymes. The transmembrane domain forms an anion exchange channel and consists of 13 helical segments embedded in the lipid bilayer and one nonhelical segment. Asparagine 642 is linked to complex carbohydrates, which protrude on the exterior of the red cell. They also function as receptors for Plasmodium falciparum, the most virulent malaria parasite. Peripheral Membrane Proteins Underlying the lipid bilayer is the peripheral membrane skeleton, an interlocking network of structural proteins, which plays a critical role in maintaining the shape and integrity of the red cell. The major proteins of the erythrocyte membrane skeleton are spectrin, actin, proteins 4. Spectrin Spectrin is the major constituent of the erythrocyte membrane skeleton and is present at approximately 240,000 molecules per cell. Both - and -spectrin contain tandem homologous spectrin repeats that are approximately 106 amino acids long and are folded into three antiparallel helices, A, B, and C. The spectrin subunit is a 246-kDa polypeptide consisting of 16 complete repeats, an N-terminal actin binding domain, a partial repeat near the C-terminus, and a nonhomologous phosphorylated C-terminus. The proteins consist of multiple homologous spectrin repeats of approximately 106 amino acids numbered from the N-terminal. The nucleation site indicates the initial region of interaction between and monomers to form an antiparallel heterodimer. Spectrin heterodimer self association into tetramers involves helix C of the 0 partial repeat of -spectrin and helices A and B of the partial 17 repeat of -spectrin to form a complete triple helical repeat. The core structure of the erythrocyte skeleton consists of spectrin heterotetramers, which are strong but flexible filaments. Repeats at the N-terminus of -spectrin (I domain) and the C-terminus of -spectrin (I domain) are the regions involved in heterodimer self-association to form tetramers. The interface of this tetramerization site is dominated by hydrophobic contacts supplemented by electrostatic interactions. At the opposite tail end of the spectrin tetramers, the N terminus of -spectrin binds to short F-actin filaments, which is potentiated by 4. The C-terminal domain varies in different isoforms of ankyrin, which are produced by alternative splicing of the gene. Membrane lipids and transmembrane proteins have been removed and the skeletons were extended during preparation and negative staining to reveal the structure. High-magnification image and schematic of the hexagonal lattice showing spectrin tetramers (Sp4) and hexamers (Sp6) or double tetramers (2Sp4). The ankyrin binding site is a flexible pocket formed by repeats 14 and 15 of -spectrin near the C-terminal end of the molecule. Nonrepeat sequences in spectrin provide the recognition sites for binding to modifiers, including kinases and calmodulin. The functions of spectrin are to maintain the biconcave disk shape of the red cell, regulate the lateral mobility of integral membrane proteins, and provide structural support for the lipid bilayer. This structure behaves like a reversible spring, which may contribute to the elasticity of the membrane. The C-terminal section of the regulatory domain varies in the different isoforms of ankyrin, proteins 2. This diversity is accomplished by the use of alternate first exons under the control of different promoters, and alternate initiation codons. The pro- tein consists of four domains, with the 30-kDa and the 10-kDa domains involved in binding to other red cell membrane proteins. The protein undergoes posttranslational palmitoylation and myristoylation, which suggests an interaction with the lipid bilayer. The adducin tails cap actin filaments and promote interaction of spectrin and actin. A primary deficiency of adducin in human disease has not been described; however, mice with targeted inactivation of - or -adducin suffer from compensated spherocytic anemia, suggesting that the adducin mutations may be candidates for recessively inherited hemolytic anemia. The length of the filaments is regulated by a "molecular ruler" of two rod-shaped tropomyosin molecules, which are bound along the filament, as well as by two tropomodulin molecules, which cap the filaments at the pointed ends. By using the cytoplasmic domains of embedded proteins as attachment points, the membrane skeleton not only affixes itself to the lipid bilayer but also influences the topology of the transmembrane proteins and constrains their lateral and rotational mobility. The membrane skeleton resembles a lattice-like network, with approximately 60 percent of the lipid bilayer directly laminated to the underlying skeleton. The protein is part of the membrane-associated guanylate kinase family and the kinase domain is close to the C terminus. The neck domain is responsible for oligomerization and the tail represents the major binding site for other red cell membrane proteins.
Purchase 25mg serophene with amex
Deferiprone (L-1) is an orally effective bidentate chelating agent; three molecules of deferiprone bind one iron atom women's health center peterborough serophene 25 mg low price. Its molecular weight is only 139 daltons and it is excreted almost entirely in the urine. Deferiprone administration is associated with a number of toxic effects, including gastrointestinal disturbances, arthropathy, transient increases in the serum levels of liver enzymes, and zinc deficiency. The main concern has centered on the propensity of the drug to produce neutropenia and agranulocytosis. It appears to be idiosyncratic, is more common in females, and appears to be reversible. It has been proposed that deferiprone enters cells and removes their iron and then passes the iron to desferrioxamine. It has been recommended for patients who are noncompliant with desferrioxamine, and like deferiprone, may be effective at removing cardiac iron. Its main toxicity is renal and hepatic, but it may also cause gastrointestinal hemorrhage. Oral Chelating Agents Chelation Therapy Chelation therapy instituted in a timely manner can decrease the potential morbidity caused by iron overload and prolong the life of patients with hereditary chronic iron-loading disorders such as -thalassemia major or intermedia. As oral chelating agents become more readily available, the application of chelation therapy to myelodysplastic states may broaden. Studies in experimental animals may not accurately reflect the role of micronutrients in humans. Accordingly, our knowledge of the effects of many micronutrients on hematopoiesis is fragmentary and based on clinical observations and interpretations that may be flawed. Inborn metabolic errors that affect single micronutrient pathways may shed light on specific effects of those micronutrients on hematopoiesis. Surveys conducted in developing countries suggest that vitamin A deficiency represents a public health problem among infants, schoolchildren and women of childbearing age. However, there is no known causal relationship between the two nutrients beyond both occurring in a setting of generalized malnutrition. Although vitamin A deficiency is recognized to occur in the United States, the relationship between it and anemia is not known. Evidence linking isolated nutritional deficiencies of pyridoxine, riboflavin, pantothenic acid, and niacin to anemia in patients is inconclusive. In animals, experimentally induced deficiency states are more commonly associated with hematologic abnormalities. These components are converted to pyridoxal 5-phosphate, which acts as a coenzyme in decarboxylation and transamination of amino acids and synthesis of aminolevulinic acid, the porphyrin precursor (Chap. Vitamin B6 deficiency induced in infants is associated with a hypochromic microcytic anemia. A review of more than 200 patients with acquired sideroblastic anemia reported that fewer than 7 percent showed greater than 1. Derangements in these pathways, sometimes involving anemia, are usually the result of inborn errors affecting the pathways of vitamin B6 metabolism and specific pyridoxal phosphate-dependent enzymes or inborn errors that lead to accumulation of small molecules that react with pyridoxal phosphate and inactivate it. Unlike iron-deficiency anemia, but similar to anemia of chronic disease, iron stores in the liver and marrow are increased, serum transferrin concentration usually is normal or decreased, and administration of medicinal iron does not correct the anemia. However, there is some evidence to suggest that vitamin A deficiency may result in impaired iron absorption or utilization5 and this may be mediated through effects on the expression of genes involved in the regulation of intestinal iron absorption. Riboflavin deficiency results in a decrease in red cell glutathione reductase activity because this enzyme requires flavin adenine dinucleotide for activation. Glutathione reductase deficiency, induced by riboflavin deficiency, is not associated with hemolytic anemia or increased susceptibility to oxidant-induced injury (Chap. There is also some evidence to suggest that riboflavin may exert its effects secondarily on other nutrients, such as folate and cobalamin. Megaloblastic anemia, responsive to thiamine, occurs in a childhood syndrome in association with diabetes and sensorineural deafness. There is usually profound anemia, megaloblastic changes with or without ringed sideroblasts in the marrow, and occasionally thrombocytopenia. The underlying defect in this condition is in the high-affinity thiamine transporter, which primarily affects synthesis of nucleic acid ribose via the nonoxidative branch of the pentose cycle. Reduced nucleic acid production through impaired transketolase catalysis appears to be the underlying biochemical disturbance that likely induces cell-cycle arrest or apoptosis in marrow cells and leads to thiamine-responsive megaloblastic anemia syndrome in these patients, which responds to lifelong administration of oral thiamine (25 to 100 mg/day). When folic acid is given to these patients in a dose of 50 mcg/day orally, a prompt hematologic response is observed. Impaired dihydrofolate reductase activity results in an inability to form tetrahydrofolic acid, the metabolically active form of folic acid (Chap. Patients with scurvy and megaloblastic anemia excrete 10-formylfolic acid as the major urinary folate metabolite. Following ascorbic acid therapy, 5-methyltetrahydrofolic acid becomes the major urinary folate metabolite. This observation has led to the suggestion that ascorbic acid prevents the irreversible oxidation of methyltetrahydrofolic acid to formylfolic acid. Under these circumstances, ascorbic acid therapy produces a hematologic response only if enough folic acid is present to interact with the ascorbic acid. Iron balance may be compromised by ascorbic acid deficiency because this vitamin serves to facilitate intestinal iron absorption by maintaining iron in a more soluble reduced or ferrous (Fe2+) state. Patients with scurvy, particularly children, may require both iron and vitamin C to correct hypochromic microcytic anemia. In patients with iron overload from repeated blood transfusions, the level of vitamin C in leukocytes is often decreased because of rapid conversion of ascorbate to oxalate. The presence of scurvy in patients with iron overload may protect them from tissue damage. Nutritional deficiency of vitamin E in humans is extremely uncommon because of the widespread occurrence of -tocopherol in food. The daily requirement of d-tocopherol for adults ranges from 5 to 7 mg, but this requirement varies with the polyunsaturated fatty acid content of the diet and the content of peroxidizable lipids in tissues. Hematologic manifestations of vitamin E deficiency in humans are limited to the neonatal period and to pathologic states associated with chronic fat malabsorption. Low-birthweight infants are born with low serum and tissue concentrations of vitamin E. When these infants are fed a diet unusually rich in polyunsaturated fatty acids and inadequate in vitamin E, hemolytic anemia frequently develops by 4 to 6 weeks of age, particularly if iron is also present in the diet. Modifications of infant formulas have all but eliminated vitamin E deficiency in preterm infants. Chronic administration of oral vitamin E 400 to 800 U/day lengthened red cell life span in some,47,48 but not all,49 studies of patients with hereditary hemolytic anemias associated with glutathione synthetase deficiency or glucose-6-phosphate dehydrogenase deficiency. Administration of vitamin E (450 U/day for 6 to 36 weeks) to patients with sickle cell anemia significantly reduced the number of irreversibly sickled erythrocytes. Cytochrome c oxidase, dopamine -hydroxylase, urate oxidase, tyrosine and lysyl oxidase, ascorbic acid oxidase, and superoxide dismutase (erythrocuprein) are cuproenzymes. More than 90 percent of copper in the blood is carried bound to ceruloplasmin, an 2-globulin with ferroxidase activity. Copper, in the form of hephaestin,54 converts iron to the ferric (Fe3+) state for its transport by transferrin. Copper deficiency has been described in malnourished children,55 and in both infants and adults receiving parenteral alimentation. These abnormalities include osteoporosis, flaring of the anterior ribs with spontaneous rib fractures, cupping and flaring of long-bone metaphyses with spur formation and submetaphyseal fractures, and epiphyseal separation. Copper deficiency with resultant microcytic anemia can be produced by chronic ingestion of massive quantities of zinc. This has been reported in patients using excessive quantities of zinc-containing dental fixatives. Despite these limitations, a serum copper level less than 70 mcg/dL (11 mol/L) or ceruloplasmin level less than 15 mg/ dL after age 1 or 2 months can be regarded as evidence of copper deficiency. In later infancy, childhood, and adulthood, serum copper values should normally exceed 70 mcg/dL. Low serum copper values may be observed in hypoproteinemic states, such as exudative enteropathies and nephrosis, and Wilson disease. In these circumstances, a diagnosis of copper deficiency cannot be established by serum measurements alone but requires analysis of liver copper content or clinical response after a therapeutic trial of copper supplementation. Copper-deficiency anemia and neutropenia are quickly corrected by administration of copper. Treatment of copper-deficient infants consists of administration of approximately 2.
