

Purchase celecoxib overnight
Typically arthritis in the back ribs order celecoxib master card, a patient will be aware of having the skin condition psoriasis long before the associated arthritis occurs. In just 15% to 25% of patients will the arthritis manifest simultaneously or subsequently (1,2). PsA can occur in up to 30% of patients with psoriasis, depending on method of ascertainment and severity of psoriasis (see Chapter 8A). Although this review will focus on pharmacotherapy of PsA, it must be recognized that optimal therapy also comprises nonpharmacotherapy approaches, including patient and family education about the disease process and therapy, exercise, nutrition, psychological counseling, physical and occupational therapy, and orthopedic surgery. A key role for the rheumatologist and rheumatology office staff is to serve as a central triage point for such adjunct therapy. These measures have been shown to effectively assess peripheral joint and skin symptoms and signs, function, quality of life, and fatigue, as well as distinguish treatment from placebo. Approaches to assessment of enthesitis, dactylitis, and spine involvement are still in development. Etanercept, infliximab, and efalizumab have been approved in the United States and Europe for psoriasis and alefacept has been approved for use in the United States. The first biologic agents approved in the United States were the T-cell modulatory agents alefacept and efalizumab, based on the key role played by T lymphocytes in psoriasis pathogenesis (26). Both block T-cell stimulation; alefacept promotes apoptosis of memory T cells and efalizumab inhibits migration of lymphocytes to the site inflammation. Both show clinically meaningful reductions in skin lesional activity and improved quality of life. When psoriasis clears it does not leave residual damage, so dermatologists typically treat till clear and then withdraw therapy until lesions return. A number of strategies have been developed for intermittent as well as combination therapy, based on assessment of skin lesion severity, to achieve optimal results (17). It is important to take into account previous tolerability and effectiveness of systemic medicines used for psoriasis when considering therapeutic options for inflammatory arthritis when it develops. Typically a patient may have already tried an over-thecounter formulation, such as ibuprofen and naprosyn, so will have a sense of their relative effectiveness and tolerability. Nonsteroidal anti-inflammatory drugs have been found to be efficacious for the treatment of spinal pain in ankylosing spondylitis, on which evidence it is reasonable to extrapolate efficacy in management of PsA spondylitis (28). Results tend to be short lived, thus of limited long-term use if inflammation is recurrent in that site. However, if the inflammation is transient in that site, then local injection therapy can be quite helpful. Systemic glucocorticoids should be used more judiciously than in other inflammatory arthritides because of the chance that psoriasis skin lesions will severely flare upon withdrawal of therapy (27). Those that are considered traditional include the oral agents methotrexate, sulfasalazine, and cyclosporine. Injectable and oral gold and azathioprine would also be considered in this group, but have been infrequently used and, in the case of injectable gold therapy, have generally fallen out of favor. Significant elevation of liver tests or drop in blood counts should lead to adjustment of dose or cessation of therapy. In the largest of these, 221 PsA patients were treated with sulfasalazine, 2 g/day, for 36 weeks (37). Although a composite arthritis score showed statistically significant improvement in the treatment group, the only individual measure within the responder index to do so was the patient global assessment, indicating that the effect was not strong. Further, there was no benefit to the skin and gastrointestinal intolerability was an issue. In 1984, Willkens published a small controlled trial using dosages of the drug that were then considered potentially appropriate in the treatment of inflammatory arthritis, 7. In this trial, only the physician global assessment of arthritis showed statistically significant improvement, and not the tender and swollen joint count. Originally placebo patients achieved a similar degree of effectiveness in joints and skin as well as inhibition of further structural damage (45). Presence of dactylitis and enthesitis, assessed by palpation of the Achilles tendon and plantar fascia insertions, decreased significantly in the infliximab group (46). Mean improvement in enthesitis and dactylitis was greater for patients receiving adalimumab, but this result did not achieve statistical significance. Radiographic progression of disease was significantly inhibited by adalimumab, as evaluated by x-rays of hands and feet, using a modified Sharp score (48). Spine disease was not assessed in these trials, due to variability of expression of this domain in this patient group. It is unknown if the same holds true in PsA, although extrapolation of this experience to PsA seems reasonable. Their efficacy in joint disease activity, inhibition of structural damage, function, and quality of life are similar. There may be some differentiation in efficacy in the skin and enthesium, but all have excellent effects in these domains. These agents tend to be well tolerated and patients generally acclimate to their parenteral administration, especially when they experience significant efficacy. Anecdotally, a similar experience has been noted in the management of PsA patients. It interferes with activation of T lymphocytes and migration of cells to the site of inflammation. It is administered subcutaneously, once per week and is approved for use in psoriasis (22). Because this response was not statistically significant, it is not recommended for treatment of arthritis (56). Traditional immune-modulating drugs can beneficially affect many of these domains as well. Observation of the effectiveness of these agents has helped elucidate the pathogenesis of PsA and psoriasis which, in turn, may lead to more novel and effective interventions. Mild disease in the joints and skin can be treated with anti-inflammatories and topical treatments. Development of targeted therapies has also increased interest in the accurate diagnosis and assessment of PsA, which facilitates the institution of appropriate therapy in a timely fashion. The treatment of psoriasis and psoriatic arthritis: an interdisciplinary approach. Assessment of patients with psoriatic arthritis: a review of currently available measures. Quality-of-life issues in psoriasis and psoriatic arthritis: outcome measures and therapies from a dermatological perspective. Traditional therapies in the management of moderate to severe chronic plaque psoriasis: an assessment of the benefits and risks. Mechanisms of action of topical therapies and the rationale for combination therapy. Long-term safety and efficacy of Adalimumab in the treatment of moderate to severe chronic plaque psoriasis. Randomized, double-blind, placebo controlled trial of low-dose pulse methotrexate in psoriatic arthritis. Long-term methotrexate therapy in psoriatic arthritis: clinical and radiological outcome. Monitoring liver function dur ing methotrexate therapy for psoriasis: are routine biopsies really necessary A randomised, double blind, placebo controlled, multicentre trial of combination therapy with methotrexate plus cyclosporin in patients with active psoriatic arthritis. Efficacy and safety of leflunomide in the treatment of psoriatic arthritis and psoriasis. Etanercept treatment of psoriatic arthritis: safety, efficacy, and effect on disease progression. Continued inhibition of radiographic progression in patients with psoriatic arthritis following 2 years of treatment with etanercept. Current evidence for the management of ankylosing spondylitis: a systematic literature review for the asas/eular management recommendations in ankylosing spondylitis. Improvement in health utility in patients with psoriatic arthritis treated with adalimumab (Humira). Alefacept treatment in psoriatic arthritis: reduction of the effector T cell population in peripheral blood and synovial tissue is associated with improvement of clinical signs of arthritis. An international, randomized, double-blind, placebo-controlled phase 3 trial of intramuscular alefacept in patients with chronic plaque psoriasis.
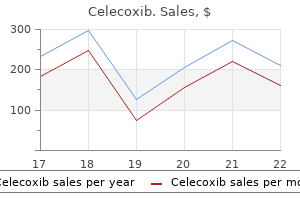
Order celecoxib american express
This may account for pathology in one testis impacting the function of the contralateral testis arthritis pain hip relief purchase online celecoxib, which has been reported with varicoceles and testicular tumors. The genital branch of the genitofemoral nerve primarily supplies sensation to the parietal and visceral tunica vaginalis and the overlying scrotum. These nerves ramify within the tunica albuginea, but do not enter the seminiferous tubules. Blood-TestisBarrier the fluid passing from the seminiferous tubules and exiting from the testis has been found to have a substantially different fluid composition than that of blood plasma or lymphatics. This suggests that compounds do not freely diffuse to and from the tubules, indicating that a barrier exists, which is known as the blood-testis barrier (Setchell and Waites, 1975). There are extremely strong, tight junctions between Sertoli cells, which provide an intracellular barrier that allows for spermatogenesis in an immune privileged site. An anechoic area between the echogenic scrotal wall and testicle is commonly visualized, which represents a small amount of physiologic fluid between the visceral and parietal layers of the tunica vaginalis. The mediastinum testis is visualized posteriorly as an echogenic band parallel to the epididymis. The echo pattern of the normal testis is fine, uniform, with a medium-level echo pattern. Color Doppler can identify testicular vessels in the majority of patients (Spirnak and Resnick, 2002). Waveforms from intratesticular arteries and testicular capsular arteries demonstrate consistently low-impedance patterns with high levels of diastolic flow. Supratesticular arteries are also sonographically identifiable and show low-impedance waveforms from the testicular, deferential, and cremasteric arteries (Middleton et al, 1989). Ultrasonography Ultrasonography is the primary imaging modality used to interrogate the scrotum and its contents. A,Onetothreeseminiferoustubules fill each compartment and drain into the rete testis in the mediastinum. The epididymis is tightly coiled and encapsulated within the tunica vaginalis sheath and would measure 3 to 4 m in length if stretched out (Von Lanz and Neuhaeuser, 1964; Turner et al, 1978). Septa form by extensions of the tunica vaginalis sheath into interductal spaces that divide the duct into histologically characteristic areas (Kormano and Reijonen, 1976). The three areas are characterized as the caput (head), the corpus (body), and the cauda (tail) of the epididymis. The tightly coiled duct, which makes up the epididymis, is continuous with the vas deferens at the most distal portion of the cauda epididymis. The duct becomes more narrow and concentric near the junction with the ductus epididymis. The diameter of the duct enlarges and becomes irregular in shape in the cauda epididymis. A cystic body on the upper pole of the caput epididymis, which may be pedunculated or sessile, is known as the appendix of the epididymis. MicroanatomicArchitecture There are two primary types of cells throughout the epididymis: principal cells and basal cells (Holstein, 1969; Vendrely, 1981). From caput to cauda, the height of the epithelium decreases, whereas the diameter of the ductus and lumen increases. There are stereocilia that shorten progressively from the proximal to the distal epididymis. The principal cells contain elongated nuclei that are commonly clefted, and they contain one or two nucleoli. As the principal cells have absorptive and secretive functions, the apex of each of these cells contain multiple coated pits, membranous vesicles, multivesicular bodies, micropinocytic vesicles, and an extensive Golgi apparatus (Vendrely and Dadoune, 1988). There is a much larger number of principal cells in the epididymal epithelium than the number of basal cells that exist there. LymphaticSupply Similar to that of the testis, the caput and corpus epididymis have their lymphatic drainage through channels that travel with the internal spermatic vein, draining to the preaortic nodes. Lymphatic channels from the cauda epididymis join those leaving the vas deferens to drain ultimately into the external iliac nodes. NerveSupply the superior portion of the hypogastric plexus and the pelvic plexus yield the intermediate and inferior spermatic nerves, respectively, which innervate the epididymis (Mitchell, 1935). Fibers from the sympathetic nervous system sparsely innervate the proximal portion of the epididymis as well as the ductuli efferentes (Baumgarten and Holstein, 1967; Baumgarten et al, 1971). The corpus epididymis includes sparse numbers of nerve fibers, and the density of nerve fibers increases progressively traveling toward the cauda epididymis. The density of fibers begins to increase at the midcorpus of the epididymis and the progressive increase in fibers is associated with the progressive proliferation of smooth muscle cells (Baumgarten et al, 1971). The basal cells are believed to be derived from macrophages and to be precursors of principal cells. There is a fair amount of variability in the nature of the epithelium of the epididymis, which is dependent on the region. There is a clear transition from a low to a high cuboidal epithelium where the rete testis and ductuli efferentes meet. The ductuli efferentes contain ciliated and nonciliated cells and the epithelium appears uneven (Holstein, 1969). The epithelium of the proximal ductuli efferentes primarily consists of nonciliated cells with extending apices thought to be for secretory function. The ciliated cells conduct sperm cells from the efferent duct to the epididymis, and they are widely dispersed throughout the epithelium (Vendrely, 1981). Junctional complexes join ciliated and nonciliated cells together at their apices, suggesting a bloodepididymis barrier (Suzuki and Nagano, 1978; Turner, 1979; Hoffer and Hinton, 1984). Nexuslike junctions connect these contractile cells to one another and each cell contains myofilaments. These cells are larger and appear like thin smooth muscle cells in the distal corpus epididymis, where they have fewer intracellular junctions. The cells have a longitudinal orientation in the two outer layers and a circular orientation in the central layer. The thickness of the distal contractile layer progressively increases as it forms the vas deferens. Ultrasonography the epididymis can be visualized ultrasonographically in its posterolateral position to the testis. The epididymis appears either hyperechoic or isoechoic in comparison to the testis (Spirnak and Resnick, 2002). Compared to the testis, the caput epididymis is typically isoechoic, the corpus epididymis is hypoechoic, and the vas deferens is anechoic (Puttemans et al, 2006). The epididymis is typically homogeneous, with well-defined echoes surrounding the epididymis that represent the fascial lining (Black and Patel, 1996). By sonographic measurement, the normal caput epididymis diameter measures between 10 mm and 12 mm and the normal corpus epididymis measures between 2 mm and 5 mm (Pezzella et al, 2013). In 98% of men, the caput epididymis is above the upper pole of the testis, with the corpus epididymis typically lateral to the testis. The epididymis is inverted with the caput epididymis inferior to the lower pole of the testis in 2. The appendix epididymis can be identified as an isoechoic structure attached to the caput epididymis (Black and Patel, 1996). Vascular flow is detectable with pulsed Doppler and color Doppler in all regions of the epididymis in nonpathologic states. ArterialSupply A branch of the testicular artery supplies the caput and corpus epididymis. This arterial branch then further divides to supply the superior and inferior epididymal branches (Macmillan, 1954). As with the testis, the deferential and cremasteric arteries also supply the epididymis and can compensate for a ligated testicular artery. The connective tissue sheaths forming septa in the epididymis are the entry points for arterial supply within the epididymis.
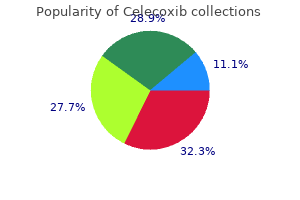
Buy celecoxib with paypal
In stark contrast arthritis pain homeopathy buy celecoxib line, the presence of large variations in chromosome numbers and complex structural rearrangements as well as intratumoral variation in these aberrations are hallmarks of most human solid tumors, which represent the bulk of human malignancies. In prostate cancer as well as most human tumor types and transplantable tumor models, the extent of chromosomal abnormalities correlates with disease severity and aggressiveness, pointing to a role for these changes in cancer progression-spanning from premalignant lesions to localized primary tumors to metastatic disease to which patients typically succumb (Brothman et al, 1990; Lundgren et al, 1992; Sandberg, 1992; Isaacs et al, 1995; Bostwick et al, 1996). For example, in a study that used a computational method to infer aneuploidy based on gene expression data of genes that are located in adjacent chromosomal regions, greater levels of aneuploidy were found to confer worse survival (Carter et al, 2006). The chromosomal changes seen in solid tumors can be broken down into two main classes: changes in the number of whole chromosomes and changes in chromosomal structure. Numerical chromosomal alterations can be subdivided further into changes in the numbers of specific individual chromosomes, aneusomies. Possible mechanisms responsible for such numerical changes include nondisjunction, endoreduplication (an abrupt doubling of the chromosome complement without cell division), cytokinesis defects, and cell fusion events. Likewise, structural changes can be subdivided into several distinct types of chromosomal aberrations as listed in Box 19-2. More recent studies on numerous human cancers found that the genomes of each cancer have numerous mutations (on average 33 to 66 somatic mutations in solid tumors) in gene coding sequences that are predicted to alter significantly the corresponding protein products (Vogelstein et al, 2013). Comprehensive sequencing efforts coupled with statistical methods to predict the effects of individual mutations have revealed that approximately 140 genes, when mutated, can "drive" tumorigenesis. Most tumors contain only two to eight such driver mutations, whereas the remaining mutations in any particular cancer case are considered "passengers" that do not confer a selective growth advantage (Sjoblom et al, 2006; Wood et al, 2007; Vogelstein et al, 2013). Such damage includes base modifications arising from reactive chemical species and from base mismatches secondary to rare misincorporation of incorrect bases or chemical conversions. In particular, the pathway is critical for repair of oxidative lesions caused by reactive oxygen species. Cancer cells that have abrogated this response may exhibit increased resistance to such therapies. Homologous recombination involves the transfer of genetic information between sister chromatids. Use of the sister chromatid is only an option following S phase into G2, before mitosis has occurred. In this instance, broken ends are brought together and ligated, often with a loss of some nucleotides at the resulting joint. Such inherited cancer predisposition syndromes further highlight the existing link between genomic instability and cancer causation. Schematic representation of the cascade at the G2M checkpoint in response to genotoxic events such as radiation. This finding implies that, at least in a subset of cases, the rearrangement may be an early event in prostate tumorigenesis. The prognostic significance of fusion status in prostate cancer remains uncertain. The precise reasons for these conflicting results are not for genomic rearrangement, termed chromothripsis (literally meaning "chromosome shattering"), has been described that was initially extrapolated from genomic sequencing studies on cancer cells (Stephens et al, 2011). Chromothripsis is evidenced by a large number (possibly hundreds) of chromosomal rearrangements in confined chromosomal regions that have occurred after apparent shattering and rejoining of a chromosomal region in a sometimes disordered fashion. As previously mentioned, cancer-associated chromosomal changes were recognized in the 1900s by Boveri, who proposed that such abnormalities might be involved in cancer causation. Much later, Klein (1981) suggested that chromosomal rearrangements affect the expression of cancer-related genes located near the observed chromosomal breakpoints. This hypothesis has been validated over the ensuing years, in large part as a result of studies on what were observed to be consistent chromosomal changes found in hematologic malignancies and soft tissue sarcomas, eventually leading to the isolation and cloning of the resident genes involved (Nowell, 1994). Over the years, painstaking dissection of chromosomal regions that are repeatedly found to undergo alteration in specific tumor types or subtypes has led to the discovery of hundreds of individual cancer-associated genes. Typically, genomic loci that are frequently lost tend to harbor tumor suppressor genes, whereas loci exhibiting copy number gains. Gene amplifications in cancer are usually seen either as multiple small extrachromosomal copies, called double minutes, or as amplified regions within chromosomes, so-called homogeneous staining regions (Cowell, 1982). In either case, the net result is to place a known oncogenic transcription factor under the control of an androgenregulated promoter, resulting in androgen-driven expression of the fusion transcript (Wang et al, 2008a). Such approaches are currently being evaluated for potential use in noninvasive diagnostic applications. Numerous variables differ among many of these studies, including the nature of the study cohort, sample size, method of cancer detection, how the tissues were obtained, intratumoral heterogeneity in the presence of the fusion (Minner et al, 2013), how the gene fusions were detected. In most cases (approximately 80%), this finding occurs through a structural rearrangement producing an isochromosome 12p-that is, a version of chromosome 12 consisting of 2 p arms and no q (long) arm. In the remaining cases, 12p material is gained through more complex chromosomal rearrangements (Rosenberg et al, 2000; Ottesen et al, 2003; Looijenga et al, 2007). More detailed analyses have revealed amplification of specific regions on 12p, including the area 12p11-12p13. Such abnormalities are wide-ranging, affecting the genome at multiple scales, including losses and gains of entire chromosomes or chromosomal arms as well as deletions and amplifications of large and small chromosomal regions. For instance, when metaphase chromosomes of tumor cells are examined during karyotypic analysis, a bewildering array of chromosomal aberrations is typically observed, such that no two karyotypes within a given cancer cell population are exactly the same. However, within this seemingly random assortment of alterations, some changes are seen in multiple different cells and multiple tumor samples, providing a strong indication that a gene or genes located in the region undergoing recurrent alteration is involved in the pathogenesis of the disease. Over the past several decades, using ever more sophisticated and higher resolution molecular methods, many such changes have been cataloged and candidate cancer genes have been identified. In the first, inherited (germline) defects in genes that cause hereditary cancer predisposition syndromes are sought, often by performing genetic linkage analysis in affected and nonaffected family members in an attempt to find genetic loci that track in a mendelian fashion with disease status. In the second approach, various techniques are employed to discover disease-associated genes in sporadic cancers that lack a strong familial component (caused by somatic rather than germline genetic alterations). In the case of cancer-related genes identified in hereditary predisposition syndromes, one hopes that alterations of genes discovered in familial forms of the disease are also relevant to their more common sporadic counterparts. In many instances, this has been found to be the case; for example, gene abnormalities linked to certain familial forms of kidney cancer are also involved in sporadic forms of the disease. HereditaryProstateCancer Family history is one of the strongest risk factors for prostate cancer (Steinberg et al, 1990), and criteria defining a hereditary form of the disease have been established (Carter et al, 1993). Twin studies have estimated a heritable risk for prostate cancer of approximately 50% (Page et al, 1997; Lichtenstein et al, 2000). Traditional linkage analysis is well suited to identify highly penetrant genetic alterations. Although this linkage was replicated in some studies, it was not confirmed in others (Cooney et al, 1997; McIndoe et al, 1997; Eeles et al, 1998; Bergthorsson et al, 2000). The frequency of the allele was much higher in men with early-onset, familial prostate cancer (3. SporadicProstateCancer In sporadic prostate cancer, initial studies found recurrent changes involving losses of genetic material at 6q, 7q, 8p, 10q, 13q, 16q, 17p, 17q, and 18q; however, in most cases, the precise genes involved have yet to be identified (Karan et al, 2003). Changes in chromosome 8, typically loss of the p arm and gain of the q arm, or portions of these arms, are the most frequently observed genetic alterations. At least two to three separate regions are deleted on 8p, implying the existence of multiple tumor suppressor genes. The region 8p22 is commonly deleted, with frequencies of 32% to 65% reported in primary tumors and 65% to 100% in metastases. Chromosome 10 also undergoes alteration in prostate cancer, with deletions observed at 10p11. An additional six regions (three with gains, three with losses) were found to be altered in greater than 10% of advanced cancers. In agreement with earlier studies, 8p was the most frequent region of genomic loss (with a peak at 8p21. The other regions commonly lost, in decreasing order of frequency, included 13q21. In contrast to the more traditional linkage analyses of the past, the new platform is amenable to very large cohorts and is better able to detect genetic variations with small to moderate effects on disease risk (Risch and Merikangas, 1996; Jorgenson and Witte, 2007; Manolio, 2010). Losses on 16q have been observed with reported frequencies ranging from 30% to 56% of prostate cancer cases, being more commonly seen in advanced cancer and associated with a poor prognosis (Carter et al, 1990; Bergerheim et al, 1991; Suzuki et al, 1996; Elo et al, 1997; Li et al, 1999). No strong candidate gene has been identified in this region (Cooney et al, 1996a; Hyytinen et al, 2002). As discussed previously, this locus also contains the p14 and p15 genes, making it difficult to pinpoint the exact target or targets of genetic loss in this region.
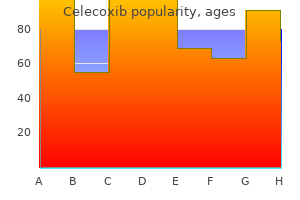
Order celecoxib 200mg without a prescription
Other factors associated with the development of tophi include early age of gout onset arthritis research discount celecoxib amex, long periods of active but untreated gout, an average of four attacks per year, and a greater tendency toward upper extremity and polyarticular episodes (9). In untreated patients, the interval from the first gouty attack to the beginning of advanced arthritis or the development of visible tophi is highly variable, ranging from 3 to 42 years, with an average of 11. Tophi may be found anywhere over the body, but occur most commonly in the fingers, wrists, ears, knees, olecranon bursa, and such pressure points as the ulnar aspect of the forearm and the Achilles tendon. Tophi also may occur in connective tissues at other sites, such as renal pyramids, heart valves, and sclerae. Similar appearing nodules are observed in other rheumatic conditions, such as rheumatoid arthritis and multicentric reticulohistiocytosis (11,12). Before antihyperuricemic agents were available, as many as 50% of patients with gout eventually developed clinical or radiographic evidence of tophi. Since the introduction of allopurinol and the uricosuric agents, the incidence of tophaceous gout has declined. Much of the knowledge about sequential development of the mature, multilobulated gouty tophus comes from the classic histopathologic descriptions of Sokoloff (13) and Schumacher (14), and the more recent immunohistochemical studies of Palmer and colleagues (15). Early-onset gout represents a special subset of cases that generally have a genetic component, show a more accelerated clinical course, and require more aggressive antihyperuricemic therapy. In large epidemiologic studies of classic gout, a family history of gout and/or nephrolithiasis is present in 25% to 30% of cases. In this younger group, detailed questioning about the kindred over several generations may yield enough information to suggest a mode of inheritance (X-linked or autosomal dominant or recessive). The heterogeneous composition of the tophus dorsal to the proximal interphalageal joint and distal phalanx is clearly revealed. The central crystalline deposit remains low intensity, but surrounding tissue enhances. The acinus has a core of noncrystalline, amorphous material surrounded by a rosette of mononuclear phagocytes. Diseases associated with overproduction of urate in children and young adults include enzymatic defects in the purine pathway, glycogen storage diseases, and hematologic disorders, such as hemoglobinopathies and leukemias. These boys, who have severe neurologic abnormalities, develop gout and kidney stones in the first decade of life if not treated early with allopurinol. Sickle cell disease, betathalassemia, and nonlymphocytic leukemias may be complicated by gouty arthritis in the young adult years. Conditions associated with uric acid underexcretion in young patients include a specific renal tubular disorder known as familial juvenile hyperuricemic nephropathy (16). This autosomal dominant disorder causes hyperuricemia from a very young age, before any evidence of renal insufficiency. The condition may lead to progressive renal failure and end-stage kidney disease by age 40. Other nephropathies associated with earlyonset gout include polycystic kidney disease, chronic lead intoxication, medullary cystic disease, and focal tubulointerstitial disease. Because organ transplant recipients use other medications, such as systemic corticosteroids and azathioprine, their gouty symptoms frequently are more atypical and less dramatic than those of patients with classic gout. Gout in Women Unlike most other rheumatic conditions, gout is less common in women than in men. In most large reviews, women account for no more than 5% of all people with gout (19). Postmenopausal gout is similar clinically in presentation and course to classic gout, except that the age of onset is later in women than in men. Conditions that are much more commonly associated with gout in postmenopausal women than with gout in men include diuretic use (95%), hypertension (73%), renal insufficiency (50%), and preexisting joint disease, such as osteoarthritis (20). Most women who develop gout before menopause have hypertension and renal insufficiency. Normouricemic Gout the most frequent explanations for apparent gout with normal levels of uric acid are that (1) gout is not the correct diagnosis or (2) the patient actually is chronically hyperuricemic but the serum urate is normal at the time it is measured (for a potential explanation of this phenomenon, see below). Several articular conditions can mimic gout closely, including crystalline arthropathies of calcium pyrophosphate dehydrate (pseudogout), basic calcium (apatite), and liquid lipid (21). Other causes of acute monoarthropathies, such as infection, sarcoidosis, and trauma, also should be considered (22). The clinical suspicions of gout should be confirmed by crystal analysis of synovial fluid. Misunderstanding the definition of hyperuricemia also can contribute to misdiagnosis of normouricemic gout. In fact, as many as one third of people presenting with acute gout to have a serum urate below Gout in Organ Transplantation Patients Hyperuricemia develops in 75% to 80% of heart transplant recipients who routinely take cyclosporine to prevent allograft rejection (17). A slightly lower frequency (approximately 50%) of kidney and liver transplant recipients develop hyperuricemia, presumably because lower doses of cyclosporine are used in these individuals. Whereas asymptomatic hyperuricemia progresses to clinical gout in only 1 in 30 subjects in the general population, cyclosporine-induced hyperuricemia leads to gout in 1 in every 6 patients (18). Other differences between primary and cyclosporine-induced gout include the marked shortening of the asymptomatic hyperuricemia and acute intermittent gout stages, with the rapid appearance of tophi. The stage of asymptomatic hyperuricemia lasts for 20 to 30 years in classic gout, but is present for only 6 months to 4 years in cyclosporine-induced disease. Normalization of serum urate values during acute gouty flares may be more common in alcoholics than in nondrinkers. In most of these cases, hyperuricemia eventually returned, although several patients with very mild gouty symptoms remained normouricemic over a prolonged period. When synovial fluids are balanced for urate concentrations, the fluids from gouty patients have a far greater propensity for promoting crystal formation than similar fluids from people with osteoarthritis or rheumatoid arthritis. A number of synovial fluid proteins have been reported to function as promoters or inhibitors of crystal nucleation. The current list of physiologically important nucleators is short, with the leading contenders being type I collagen and a gamma globulin subfraction (10). The degree of hyperuricemia correlates positively with the overall risk of acquiring gout. However, rapid increases or decreases in the concentration of synovial fluid urate are related more closely to actual precipitation of the acute gouty attack. A rapid flux in urate level is a triggering mechanism in gout induced by trauma, alcohol ingestion, and drugs. The trauma may be as minor as a long walk and may not have caused pain during the activity, but it caused intra-articular swelling. When the joint is allowed to rest, there is a relatively rapid efflux of free water from the joint fluid. This results in a sudden increase in synovial fluid urate concentration, which may allow precipitation of urate crystals and a gout attack. The consumption of lead-tainted moonshine results in chronic renal tubular damage that leads to secondary hyperuricemia and saturnine gout (the word saturnine, meaning of or relating to lead, is derived from the belief of the ancients that this metal comprised the planet Saturn). The ingestion of any form of ethanol can raise uric acid production acutely by accelerating the breakdown of intracellular adenosine triphosphate (25). Beer consumption has an added impact on gout because it contains large quantities of guanosine, which is catabolized to uric acid (26). Thiazide diuretics selectively interfere with urate excretion at the proximal convoluted tubule. Low dose aspirin (less than 2 g/day) also can raise serum urate levels, but higher doses have a uricosuric effect and may lower the serum urate concentration. A rapid increase or reduction in the serum urate level can provoke gouty attacks; allopurinol is the drug most often responsible for this effect. The mechanism for this paradoxic response appears to be the destabilizing of microtophi in the gouty synovium when the urate concentration of the synovial fluid is changed rapidly. As the microtophi break apart, crystals are shed into the synovial fluid and the gouty episode is initiated (27). Three forms of hyperuricemia-induced renal disease are recognized, including (1) chronic urate nephropathy, (2) acute uric acid nephropathy, and (3) uric acid nephrolithiasis. Although chronic hyperuricemia is thought to be the cause of urate nephropathy, this form of kidney involvement is essentially never seen in the absence of gouty arthritis. Progressive renal failure is common in people with gout, but the attribution of renal failure to chronic urate nephropathy itself is often difficult owing to the frequent confluence of multiple comorbid conditions in patients with gout.
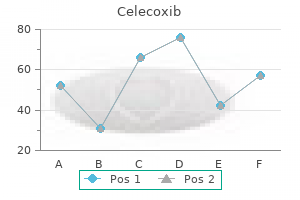
200mg celecoxib free shipping
Fibrillin is rich in cysteine arthritis swan-neck deformity buy celecoxib 200 mg visa, and intra- and interchain disulfide bonds are crucial to the formation and function of microfibrils. Approximately one half of patients respond biochemically and clinically to large doses of vitamin B6 (usually more than 50 mg pyridoxine per day), an obligate cofactor for cystathionine beta-synthase. Adequate levels of folate and vitamin B12 are required for therapeutic and biochemical response. Preexisting mental retardation and ectopia lentis are not reversed by pyridoxine treatment in patients who show biochemical correction, emphasizing the need for early diagnosis and therapy. Unfortunately, some pyridoxine responders may escape detection in the typical screening protocols. In pyridoxine nonresponders, a low methionine diet and oral betaine therapy (to stimulate remethylation of homocysteine to methionine) are the usual treatments; this approach can be successful if the diet and vitamin are tolerated. The Stickler syndrome can be caused by mutations in at least four genes, three of which have been identified (21). Variability in clinical features among families is much more extensive than within a family, which likely reflects the genetic heterogeneity. Within individual types, however, biochemical studies have demonstrated considerable heterogeneity. Extensive phenotypic and biochemical characterization nonetheless fails the clinician as often as it helps; approximately one half of patients who have at least one "cardinal" feature defy categorization. Congenital dislocation of the hips in the newborn, habitual dislocation of joints in later life, joint effusions, clubfoot, and spondylolisthesis are all consequences of loose jointedness. Hemarthrosis and "hemarthritic disability" have been described and are analogous to the bruisability of the skin in this syndrome. Many people with mild joint laxity and without joint instability are labeled as having this type, particularly if relatives show a similar manifestation (25). In some cases, such labeling does more harm than good, unless it is made quite clear that little disability, if any, is likely. The cardinal features relate to the joints and skin: hyperextensibility of skin, easy bruisability, increased joint mobility, and abnormal tissue fragility (22). Skin involvement is variable: thin, nearly translucent skin is present in some, and mildly hyperextensible skin is the only feature in others. Inheritance usually is autosomal dominant, and many cases are sporadic occurrences in their pedigree, suggesting they are heterozygous for a new mutation. Because this test is relatively straightforward to conduct, and because the results are highly specific for this condition, the diagnosis often is confirmed biochemically (27). Vitamin C is a necessary cofactor of lysyl hydroxylase, and pharmacologic doses may be beneficial. The underlying defect is an inability to cleave the N-propeptide from type I procollagen, a process that is necessary for conversion to mature collagen. Mutations at the cleavage site of alpha 1(I) and alpha 2(1) procollagens can cause this phenotype; the procollagen defects behave as dominant traits. Classification is based on inheritance pattern and clinical criteria (Table 31-2). It is autosomal dominant and is associated with considerable intrafamilial variability. One patient might be markedly short of stature, with frequent fractures and much disability, while an affected relative leads an unencumbered, vigorous life. Defects in the genes for both alpha 1(I) and alpha 2(I) procollagen can cause this syndrome. The disorder affects primarily skin and fascia, and joints are affected little (28). Recurrent dislocation, especially of the shoulder or patella, is the usual presenting complaint (29). Joint hyperextensibility is variable but usually mild, and skin involvement is uncommon. Autosomal dominant inheritance with marked variability within a family is the rule, emphasizing the need for a comprehensive family history, including examination of close relatives if possible. Larsen syndrome is characterized by congenital dislocations; a characteristic facies of prominent forehead, depressed nasal bridge, and widely spaced eyes; joint dislocations; and skeletal dysplasia. Dislocation occurs at the knees (characteristically, anterior displacement of the tibia on the femur), hips, and elbows. Cleft palate, hydrocephalus, abnormalities of spinal segmentation, and moderate-to-severe short stature have occurred in some. The recessive form is associated more often with severe short stature and neurologic complications of spinal deformity. Most cases arise as the result of a new mutation (the phenotype being dominant transmissable, if the patient lived and reproduced) in either alpha 1(I) or alpha 2(I) procollagen. A "dominant-negative" model explains the severe phenotype produced by a heterozygous mutation. Rare patients have affected siblings and normal parents; in some of these cases, a parent has been shown to have the mutation at low level in the gonads, resulting in the potential for multiple affected offspring. It usually occurs sporadically, suggesting new mutations, or autosomal recessive inheritance. Natural History the natural histories of skeletal involvement among these types bear similarities. In such cases, the limbs are likely to be short and bent at birth, and multiple rib fractures give a characteristic "beaded" appearance on radiographs. Brittleness and deformability result from a defect in the collagenous matrix of bone. Joint laxity sometimes is striking in type I; dislocation of joints can result from deformity secondary to repeated fractures, lax ligaments, or rupture of tendons. In one family, susceptibility to osteoporosis was found to be due to a mutation in type I collagen. This emphasizes that the ability to identify mutations in a particular gene does not necessarily facilitate clinical diagnosis. Furthermore common problems that are not thought to be syndromic may be due to defects in one or another of the components of the extracellular matrix. Calcium supplementation, calcitonin, and vitamin D are ineffective unless clear deficiency is present. Treatment with oral or injected bisphosphonates has had considerable success in young patients in reducing fractures and improving skeletal growth (32,33). However, the overall quality of the bone may not be improved and the issues of how long to treat children and whether to treat adults remain unresolved (34). Although phenotypically diverse, the individual disorders share mucopolysacchariduria and deposition of catabolites of proteoglycan in various tissues. The chief radiologic features are a thick calvaria; an enlarged, J-shaped sella turcica; a short and wide mandible; biconvex vertebral bodies; hypoplasia of the odontoid; broad ribs; short and thick clavicles; coxa valga; metacarpals with widened diaphyses and pointed proximal ends; and short phalanges. In these forms, progressive arthropathy and transverse myelopathy secondary to C1-C2 subluxation account for considerable disability. The latter frequently presents as obstructive sleep apnea or complications with general anesthesia. Genetic disturbances of mucopolysaccharide metabolism without mucopolysacchariduria include the mucolipidoses (42). Myocardial infarction, stroke, and gastrointestinal hemorrhage are complications of vascular involvement and the leading causes of death. The condition gets its name from characteristic skin lesions that develop in regions of flexural stress and resemble plucked chicken skin. This raises the likelihood that the effects on the extracellular matrix are due to some circulating metabolite that is dysregulated because of the transport defect (38). An inexorable course begins in the first year or so of life, usually with a seemingly inflammatory process and nodule formation on the back of the thorax, neck, or scalp.
Order 100mg celecoxib otc
In patients from high-risk ethnic groups with typical symptoms and a therapeutic response to colchicine rheumatoid arthritis with rheumatoid factor discount celecoxib uk, genetic testing is usually not considered necessary to confirm the diagnosis. Treatment Daily oral cochicine therapy is the mainstay of treatment; it prevents both acute attacks and the development of amyloidosis. Diarrhea is a common side effect of colchicine, but may be minimized by starting at a low dose and gradually titrating upwards, by dividing the daily dose, and by taking appropriate measures should lactose intolerance develop. Neuropathy and myopathy are rare complications observed mainly in the elderly and those with renal impairment. There may be a slightly increased risk of trisomy 21 in the offspring of parents taking colchicine at the time of conception. The use of intravenous colchicine to abort acute episodes may cause severe toxicity in those taking daily oral colchicine. Clinical and Laboratory Findings the first attack usually starts in infancy, often precipitated by childhood immunizations. Episodes may occur once or twice a month in childhood and adolescence, and may become less frequent or severe when the patient reaches adulthood. A number of cutaneous manifestations have been described, including diffuse painful erythematous macules, urticaria, and a morbilliform rash. Large joints are usually affected, synovial fluid shows a predominance of granulocytes, and x-rays do not usually show erosions. It is likely that this latter category represents an etiologically heterogeneous group of patients. Other subsequently identified mutations have been shown to interfere with hydrogen bonding or to add or delete amino acids in the extracellular domains of the receptor. Although there is a great degree of variability, attacks may last more than 1 month at a time. The risk of amyloidosis may be somewhat higher among those with a positive family history of amyloidosis and those with mutations causing substitutions at cysteine residues. Treatment Daily colchicine neither prevents acute attacks nor prevents the development of amyloidosis. Patients on corticosteroids frequently require increasing doses over the course of their illness with attendant increased adverse effects. Clinical and Laboratory Features Patients with the cryopyrinopathies usually present very early in life with fever, an urticarialike skin rash, and an intense acute-phase response. The rash is not true urticaria, in that there are infiltrates of granulocytes and lymphocytes rather than mast cells. The severity of joint and neurological involvement, along with the risk of amyloidosis, helps to distinguish among the three clinical disorders, although there is considerable overlap. Familial cold autoinflammatory syndrome has a clear episodic quality, with attacks of rash, fever, polyarthralgia, and constitutional symptoms occurring 1 to 2 hours after generalized cold exposure. Chronic aseptic meningitis also occurs, which can lead to headache, increased intracranial pressure, and intellectual impairment. Sensory organ involvement includes sensorineural hearing loss, conjunctivitis, and uveitis, sometimes leading to deafness and/or blindness. Untreated, about 20% die before reaching adulthood, and amyloidosis may occur later in life. Nevertheless, the availability of genetic testing has markedly increased the awareness of these disorders. Vision remained stable in all patients, and hearing actually improved in one third. Complete remission of inflammation occurred in 10 of the 18 symptoms after 6 months of treatment. Magnetic resonance imaging demonstrated marked reduction in cochlear and leptomeningeal enhancement. The mean age of onset is approximately 45 years, with a relatively even gender balance. The joints affected include the interphalangeal joints of the hands and feet, the wrists, shoulders, and ankles (22). Tender periarticular swelling, 2 to 4 cm in diameter, may accompany an attack, or occur independently. Small, sometimes painful subcutaneous nodules may develop near the elbows, wrists, or knees, with a particular predilection for the fingers. Synovial biopsies and fluid taken during an attack demonstrate polymorphonuclear leukocytes. Biopsies of subcutaneous nodules demonstrate inflammatory cells, and notably lack the areas of fibrinoid necrosis and palisading mononuclear cells seen in rheumatoid nodules. Intermittent Hydrarthrosis Intermittent hydrarthrosis is characterized by periodic episodes of monoarticular or pauciarticular arthritis. Attacks are notable for the periodicity of their nature, such that patients can accurately predict their next attack. The usual age of onset is between 20 and 50, with a relatively even gender balance. There are cases in which attacks began at menarche, coincided with menses, and remitted during pregnancy and following menopause. Clinical and Laboratory Features Attacks involve episodes of pain, swelling, and limitation of movement, usually affecting a single joint, although occasionally more than one joint may be affected. In most cases, attacks last 3 to 5 days, with massive joint effusions but no erythema or warmth. In an individual patient, a limited number of joints may be affected, the most commonly affected being the knee, with the hip, ankle, and elbow less frequently involved. Radiographs demonstrate soft tissue swelling but no erosions, even in patients with repeated attacks. Mutation and haplotype studies of familial Mediterranean fever reveal new ancestral relationships and evidence for a high carrier frequency with reduced penetrance in the Ashkenazi Jewish population. Simvastatin treatment for inflammatory attacks of the hyperimmunoglobulinemia D and periodic fever syndrome. Eosinophilic Synovitis this rare condition is described in individuals with a history of atopy. Both genders are affected equally, and the typical age of onset is between 20 and 50 years (25). Eosinophilic synovitis has been proposed to be the synovial equivalent of dermatographism. It has been speculated that trauma may trigger activation of mast cells, attracting eosinophils and thus producing an effusion (25). Episodes are self-limiting, lasting up to 2 weeks, but require only symptomatic treatment. Clinical and Laboratory Features Swelling develops rapidly, usually over 12 to 24 hours, and lasts for 1 to 2 weeks. Although the effusions are large, there is little associated pain, warmth, or erythema. The peripheral white cell count, and in particular the eosinophil count, is normal, although some patients have elevated IgE levels. Synovial fluid shows mildly elevated leukocyte counts, with 16% to 52% eosinophils in the initial series (25). Synovial eosinophilia is also seen in patients with metastatic adenocarcinoma and following arthrography. Features suggestive of eosinophilic arthritis, in contrast to the wide differential mentioned, include a personal or family history of allergy and dermatographism (25). Palindromic rheumatism: part of or apart from the spectrum of rheumatoid arthritis. Prognostic factors for the development of rheumatoid arthritis and other connective tissue diseases in patients with palindromic rheumatism.
Generic celecoxib 200mg line
In one study socks for arthritic feet buy online celecoxib, one fourth of both disease groups lost greater than 1 height Z score over the 14-year follow-up period (1). The greatest effect on velocity was seen in children with more severe polyarticular disease. Sixty-nine percent of youth with polyarticular and systemic disease had orthodontic abnormalities (6). Polyarticular patients often have small, short facies with underdeveloped mandibles. Corticosteroid injection may be helpful in selected patients and costochondral grafts have been used in severely affected patients. Both the cortical appendicular skeleton and the axial trabecular bone are affected, but the cortical to a greater degree (7). Other factors, including decreased physical activity, immobility, decreased sun exposure, and decreased dietary intake of calcium and vitamin D, are additional contributing factors. Peak bone mass is normally reached during adolescence and this achievement is important to minimize future risk for osteoporosis and fractures. Significant axial osteopenia of lumbar spine and femoral neck was found in patients with polyarticular disease (8). Therapy includes weight-bearing exercise, appropriate nutrition, calcium and vitamin D supplementation, and, most importantly, adequate disease control with suppression of inflammation. Early study of bisphosphonate therapy in children with rheumatic disease has been encouraging but not without adverse effects (10). In addition to generalized osteopenia, involved joints often show local juxta-articular demineralization even on early radiographs. Local Local growth disturbances occur as a result of inflammation and the accompanying increase in vascularity, which may result in either over- or undergrowth of the affected bone. The small size was thought to be secondary to destruction of the articular cartilage (4). The distal femoral epiphysis accounts for approximately 70% of femoral growth, so persistent inflammation leads to overgrowth on the involved side in a child whose epiphyses have not yet closed. Osteocalcin levels in patients with heights less than the third percentile were below normal, suggesting decreased osteoblast activity (13). However, these patients were also on corticosteroid therapy, which can decrease osteocalcin levels. However, at the end of 1 year the height velocity decreased to pretreatment levels (18). Fifty percent of the children in this study had borderline or poor caloric intake (13). Growth hormone therapy may be beneficial in some patients but the response is unpredictable. Insulin-like Growth Factor 1 Insulin-like growth factor 1 is a peptide produced in the liver and is the main periperal mediator of growth hormone. Levels appear to correlate with the degree of inflammation as measured by acute phase reactants. In a separate study, antithyroid antibodies were found in a higher frequency in children with arthritis, especially pauciarticular disease, than in the general population (21). These findings suggest that careful monitoring of thyroid function in children with arthritis is indicated. Using anthropometric measurements, up to 40% have poor nutritional status and muscle mass is frequently low. Protein stores as well as specific nutrients such as iron, selenium, vitamin C, and zinc have been reported as low (23). In a recent study, undernutrition was present in 16% of the children with arthritis, including those with pauciarticular disease (24). In addition, some patients have mechanical feeding problems related to jaw or upper extremity disease. Vascular Endothelial Growth Factor this factor is a mitogen for vascular endothelial cells and a mediator of vascular permeability. Dietary logs, nutrient analysis, and consultation with a dietitian is needed for a child with continued poor weight gain. Reported incidence varies from 5% to as high as 50%, but recent studies show an incidence of 12% to 25% (25,26). Current guidelines for frequency of ophthalmologic examination are available (27). As uveitis can develop after the onset of arthritis, ongoing monitoring is important. Long-term outcome for adult patients with childhood-onset uveitis is still poor, with visual acuity impaired in 40%, poor in 20%, and lost in 10% (28). Therefore, the outcome of uveitis with onset in the past 5 to 10 years will likely be much better than the existing reports (see Chapter 7A for more information) (29). The consequences of nonadherence are multiple, not only for the patient but also for the health care system. Poor or dishonest communication between patient and physician or health care provider also stresses the relationship and may lead to needless changes of medication or unnecessary testing. These can generally be grouped into three categories: (1) factors relating to the disease; (2) factors related to the patient and family; and (3) those related to the regimen itself. There is no typical noncompliant patient and no consistent correlations with obvious demographic factors. However, certain states that lead to noncompliance have been reported (33) and are included in Tables 7D-1 and 7D-2. Strategies often involve a coordinated list of activities, including taking regular medication, complex exercise regimens, dietary modifications, regular clinic visits and laboratory tests for monitoring the patient, and, in some children, wearing of therapeutic splints. This is complicated by the fact that there is often delayed benefit for good compliance. In fact, estimates are that only 50% to 54% of patients with chronic pediatric disease adhere adequately with their recommended therapy (30). Surprisingly in the prednisone study, two thirds of patients overmedicated themselves, possibly when they felt poorly. Adherence with exercise regimens are likely to be lower than with medication, ranging from 47% to 67% by parent report (33). Negative reactions from the child, including complaints, refusal, discomfort, or embarrassment, or more general oppositional behavior. Lack of understanding of the disease and treatment, especially in younger children. Duration, often prolonged with unpredictable exacerbations- compliance tends to decrease over time. When a patient is asymptomatic or in remission, there is often a temptation to discontinue medication because the patient feels well. This is enhanced by a commonly seen delay in therapeutic response of days to weeks and also by the delay in occurrence of negative effects (recurrence of symptoms) with missed medication. Exercise regimens may be especially problematic because the child may experience discomfort and express anger or resentment toward the parent. Children with chronic disease may be asked to alter their lifestyle in a way that restricts their sports interests or peer-related social activity or reduces their leisure time. Parental supervision and appropriate involvement are critical in providing the children with needed support. Delay in receiving or lack of subspecialty care and implementation of appropriate therapeutic regimens may also contribute to poor functional outcomes. Organizational strategies include counseling, increasing supervision, decreasing complexity, decreasing costs, and increasing palatability of medication. Behavioral strategies can include self-management training to increase self-esteem, training for parents to deal with oppositional behavior, and monitoring adherence and using reinforcement or a reward system for good adherence. Reinforcement programs are time consuming and require parental training but can improve adherence (33). Regular clinic visits are important to re-educate and reinforce adherence strategies and adapt treatment. They also help build and maintain a cooperative and trusting relationship between clinician and patient. The clinician must relate to the child, who needs to be an active partner in his or her treatment program. Further studies are needed to identify children and families at high risk for nonadherence, further define successful family coping mechanisms that can be taught and reinforced, and to evaluate strategies for improving compliance.
Celecoxib 100mg overnight delivery
Osteotomy Proximal femoral osteotomies arthritis pain vinegar order celecoxib 100mg otc, technically challenging to perform, have shown poor-to-moderate success with the exception of studies in Japan. This difference may be in part due to differences in the pattern of vascular anatomy found in the specific patient populations. The objective of an osteotomy is to redistribute forces to healthy bone by moving necrotic tissue away from areas which are weight-bearing. To increase the effectiveness of this procedure, surgeons have begun utilizing bone growth factors. By fitting the prosthesis in this manner, the chance for dislocation following surgery decreases. This procedure also conserves bone stock and readily allows for conversion to a total hip replacement in the future. Metal-on-metal resurfacings are more comprehensive than limited resurfacings, providing better functional results, more effective pain relief, and improved range of motion. First introduced in the middle of the last century, metal-on-metal resurfacings were discounted initially as a viable arthroplasty because of the unacceptably high number of cases with component loosening and failure. New technology creating improved bearing surfaces has triggered renewed interest in this technique. Nonvascularized Bone Grafting the purpose of bone grafts is to provide structural support to the subchondral bone and articular cartilage. The graft may be inserted through a trap door window in either the femoral head or neck. Surgeons have also begun to use growth and differentiation factors to improve the outcome of this procedure. Vascularized Bone Grafting Vascularized bone grafting was suggested as an alternative to nonvascularized bone grafting after initial results suggested there was not adequate vascularization following the procedure. Similar to nonvascularized bone grafting, growth factors and osteogenic factors may increase the effectiveness and improve the long-term outcome of vascularized bone grafting. The downside to this procedure is that it is technically difficult, timeintensive, and associated with higher donor-site morbidity. Furthermore, this procedure requires two teams: one team must prepare the femur while the other harvests the fibula. Total joint arthroplasty and other major surgical procedures are likely inappropriate for patients with chronic disease or short life expectancies. Because of their increased life expectancy, younger patients are more likely to require a revision. In the future, more nonoperative methods may be available for successful hip preservation. Osteoarthritis-like disorder in rats with vascular deprivation-induced necrosis of the femoral head. Risk period for developing osteonecrosis of the femoral head in patients on steroid treatment. Core decompression for avascular necrosis of the distal femur: long term followup. Core decompression of the femoral head for osteonecrosis using percutaneous multiple small-diameter drilling. Use of metal-on-metal total hip resurfacing for osteonecrosis of the femoral head: an analysis of 42 hips compared to osteoarthritis. Statin therapy decreases the risk of osteonecrosis in patients receiving steroids. The use of alendronate to prevent early collapse of the femoral head in patients with nontraumatic osteonecrosis. The management of necrosis-associated and idiopathic bone-marrow oedema of the proximal femur by intravenous iloprost. Bone pain, fracture, and nerve impingement can occur due to enlarging and poorly constructed bone matrix. Potent and generally safe suppressive agents have been developed and have resulted in a more aggressive approach to therapy. Initially, there is active resorption by large and increased numbers of osteoclasts containing multiple nuclei, followed by deposition of bone by numerous osteoblasts which most often results in a weakened disorganized bony structure that is interspersed with areas of fibrosis. Although localized to isolated areas of the skeleton, there may be widespread bony distribution. The bones most commonly affected are the pelvis, femur, skull, tibia, vertebrae, clavicle, and humerus. There is additional evidence suggesting a virus trigger (from the paramyxovirus family) (3). Vertebrae may enlarge, weaken, and fracture, resulting in a loss in body height and a stooped forward (or simian) posturing. Involved vertebrae may compress nerves from the spinal cord, resulting in pain, dysesthesias, weakness, or even lower extremity paraparesis or paraplegia. Long bones become bowed, resulting in reduced function, abnormal gait, and contractures. The diagnosis is uncommonly suspected on the basis of symptoms and physical examination. The extent of bony involvement can be determined from a pertechnitate radionuclide bone scan. Suppressive therapies are not expected to correct existing hearing loss, deformity, or osteoarthritis. Bed rest should be avoided, if possible, to prevent hypercalcemia and immobilization osteoporosis. Surgery is often helpful in removing pressure from compressed nerves or to replace a secondary osteoarthritic joint. Other agents such as parenteral calcitonin and plicamycin are still occasionally of value. Adverse effects that are common to the bisphosphonates include esophagitis and bone pain. Mandibular osteonecrosis has been reported with all bisphosphonates, but is particularly uncommon, and is most common with high-dose intravenous bisphosphonates (5). Alendronate (Fosamax, Merck) is a potent nitrogencontaining bisphosphonate administered orally as 40 mg/day for 6 months (6). Alendronate can be administered for 3 months if the alkaline phosphatase has normalized. Alendronate is well tolerated with esophageal and gastric ulceration as the major adverse effect. The duration of response correlates inversely with the pretreatment level of serum alkaline phosphatase. The response to repeat therapy with disodium etidronate is variable and there is increasing resistance to repeated retreatments. Non-union fractures tend to calcify their callus following disodium etidronate therapy. The dose of disodium etidronate is 5 mg/kg per day or 400 mg daily for a 40-kg (88 lb) to 80-kg (176 lb) patient. Adverse reactions to disodium etidronate include abdominal cramps, diarrhea, hyperphosphatemia, increasing bone pain, and a possible increase in fractures. Hyperphosphatemia appears to be due to a direct renal effect of disodium etidronate. Pamidronate should be administered as 60 to 90 mg intravenous infusions with normal saline or dextrose/ water. A variety of regimens have been used effectively, including daily infusions for 3 to 5 days, once weekly for 3 to 5 weeks, once with re-evaluation monthly, etc. Risedronate (Actonel Procter & Gamble/sanofiaventis) is a potent tertiary nitrogen containing bisphosphonate administered orally as 30 mg/day for 2 months (9). There are reports of esophagitis and other nonspecific gastrointestinal complaints. Tiludronate (Skelid Sanofi Winthrop Industrie) is a nitrogen-containing bisphosphonate administered orally as 400 mg/day for 3 months (10). Zoledronate (Zometa Novartis Pharmaceuticals) is a potent bisphosphonate administrated as a 4-mg intravenous 15-minute infusion. The dose can be decreased or the interval between doses increased depending on the response to therapy. Most patients will have chemical exacerbation within 6 months of calcitonin discontinuation.
