

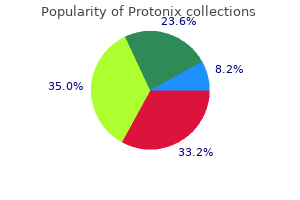
Generic protonix 20mg fast delivery
Mental activity (emotional stress and mental arithmetic) can increase sweat secretion gastritis upper right abdominal pain buy protonix from india. This sweating tends to affect the palms, axillae, and plantar areas predominantly. Thermal sweating due to heat exposure occurs over most of the body surface and is normally less prominent on palmar and plantar surfaces. The reasons for these differences are not clear but may relate to enhanced sweat gland responsiveness to a combination of factors including the influence of the central autonomic network on regional sweating and enhanced adrenergic sweating in palmar, axillary, and plantar regions. The condition can be familial and is often socially disturbing to young adults who seek treatment. Shapiro syndrome is very rare and is characterized by paroxysmal hypothermia and hyperhidrosis (due to abnormal hypothalamic thermoregulation), and agenesis of the corpus callosum. Localized hyperhidrosis may occur to compensate for sweat loss elsewhere (compensatory hyperhidrosis). Rare cases of essential hyperhidrosis occur in which the acral (distal) parts sweat heavily, whereas other body parts do not sweat. Contralateral hyperhidrosis following cerebral infarction has been described as an uncommon complication possibly due to interruption of descending inhibitory pathways. Patients with cervical and upper thoracic complete spinal cord traumatic transections are frequently troubled with localized hyperhidrosis of the head and upper trunk when noxious stimuli below the level of their lesion cause autonomic hyperreflexia. When accompanied by paroxysmal hypertension, this syndrome can be confused with pheochromocytoma and is often caused by a distended bladder or rectum. In syringomyelia, the excessive sweating is segmental and often appears in dermatomes in which sensation is later disturbed. Hyperhidrosis with partial nerve trunk injury occurs as part of a complex regional pain syndrome and may be due to an obvious lesion or an occult problem, such as paraspinal metastatic deposits affecting the sympathetic chain or segmental spinal nerve trunk. Paroxysmal, localized hyperhidrosis may occur in the coldinduced sweating syndrome and as an idiopathic disorder responsive to clonidine. Some patients who undergo endoscopic thoracic sympathectomy for palmar hyperhidrosis develop compensatory hyperhidrosis of the thorax and abdomen. Hypohidrosis and Anhidrosis Hypohidrosis is the reduction in sweating and anhidrosis is the absence of sweating. Disorders of the skin, developmental dyshidrotic syndromes (such as X-linked anhidrotic ectodermal dysplasia), lesions of the autonomic nuclei or their projections, and various pharmacological and neurotoxic agents can be responsible for the loss of sweating (sudomotor failure). Physiological hypohidrosis occurs in skin over bony prominences, in proximal extremities in the elderly, and in dehydrated states in which generalized hypohidrosis, delayed sweat onset, and a greater increase in core temperature with heat exposure are observed, especially in men. More often, patients are aware of areas of compensatory hyperhidrosis (often body segments immediately rostral or caudal or contralateral to the areas of anhidrosis) or simply do not notice a decrease in their sweating. Disorders of Thermoregulatory Sweating Conditions causing disturbed sweating can be categorized as those producing hyperhidrosis and anhidrosis. Hyperhidrosis Generalized hyperhidrosis can be primary (some cases of essential hyperhidrosis, Shapiro syndrome) or secondary and due to medical disorders such as pheochromocytoma, thyrotoxicosis, acromegaly, chronic infection, and malignancy. Essential (primary focal) hyperhidrosis is a common condition producing excessive sweating in the hands, feet, and axillae at nonelevated core temperatures. Aging leads to regional decreased sweat output causing a significant impediment to the maintenance of body temperature with passive heating. Also, there is an age-related loss of the intermediolateral cell column neurons and their decreased ability to augment sweat gland secretory capacity with aging in both men and women. This age-related autonomic failure may explain the occasional incomplete sweat patterns and delayed sweating seen in elderly who are otherwise neurologically healthy. Because the symptoms of thermoregulatory sweat loss are often overlooked, it is important to be able to objectively test for sudomotor loss on standardized tests of sweating. The thermoregulatory sweat test, which is one of the simplest and most direct ways of evaluating thermoregulatory sweating, is discussed in the following section. Thermoregulatory Sweat Test Clinical and Neurophysiological Considerations In this test, a controlled, tolerable heat stimulus is given to recruit all areas of skin capable of sweating. Afferent skin temperature thermoeffector loops and core temperature elevation are utilized to obtain a maximal sweat response. The test has been used to evaluate sweating for a long time and recently has been confirmed to provide an effective stimulus to recruiting a maximal sweat gland response. Results can also be quantitated as the percentage of body surface area not sweating. The thermoregulatory sweat test is most often used as a diagnostic tool for conditions such as the neurodegenerative disorders. The ability to examine the entire anterior body surface simultaneously for both central and peripheral sweating disorders makes this test an excellent screening test of autonomic function. The percentage of body surface anhidrosis correlates highly with the degree of symptoms and signs in diabetic autonomic neuropathy and is a useful measure to following the course of the disorder. A maximal sweat response occurs when both central (oral) and mean skin temperatures are increased in a moderately humid environment in which some degree of sweat evaporation can occur. Several techniques, including hot baths and infrared and incandescent heat lamps, have been used for the past 50 years to produce sweating, but the most satisfactory method is to use a cabinet in which the environment is controlled and the entire body (including the head) is heated. The last requirement maximizes the reflex sweat response without directly causing skin to sweat due to heat injury. The major components of the sweat cabinet are the insulated walls with windowed access doors and a transparent head end enclosure, and a regulated heated air source that has been humidified via a steam generator and overhead infrared heaters (which heat the skin and are carefully regulated by skin temperature feedback control). One powder (alizarin red S) is in orangeyellow color in the dry state and turns purple when it contacts sweat moisture. This produces a distinct, colorful delineation of anhidrotic and sweating skin that can be photographed to document the sweat distribution. Indicators currently in use include alizarin red S, starch and iodide, and iodinated soluble starch. Examples of the most commonly encountered abnormal sweat distribution patterns: Distal (5-1), Segmental (5-2 and 5-4), Regional (5-3), Global (5-6), Focal (5-5), Normal (5-7), and Mixed (5-2, 5-4, 5-5). Type 1 (heavy generalized) is a common pattern of sweating usually seen in male subjects. Type 2 normal subjects exhibit lighter sweating of proximal extremities with heavy sweating elsewhere. Type 3 subjects have lighter sweating of the lower body as well as proximal extremities. With all types, there may be sweat loss over bony prominences such as the patellae. Each exhibits an asymmetrical segmental sweat loss and segmental hyperhidrosis, the latter often more noticed by the patient (anhidrosis in yellow color). Abnormal Sweat Distributions Diabetes mellitus produces distinct peripheral neurological disorders, including length-dependent axonal neuropathy, painful truncal radiculopathy, and autonomic neuropathy. Truncal radiculopathy has a distinct clinical presentation of agonizing, and occasionally lancinating, pain associated with cutaneous dysesthesia. The percentage of anterior body surface that does not sweat (percentage anhidrosis or thermoregulatory sweat test percent) is then determined by planimeter measurements or by counting pixels with a computer. The thermoregulatory sweat test percent can serve as a useful quantitative measure of sympathetic sudomotor failure. Clinical Applications of the Thermoregulatory Sweat Test this test is helpful in identifying autonomic involvement in many other neurological disorders. An important use involves the evaluation of patients presenting with an extrapyramidal syndrome. In this situation, tests of peripheral or postganglionic sudomotor function will be intact in areas anhidrotic on the thermoregulatory sweat test. As the disease progresses, there is global thermoregulatory anhidrosis, and postganglionic sympathetic sudomotor function declines. Sweating deficits caused by peripheral neuropathy with small-fiber axonal involvement Nerve conduction Quantitation and Reporting of the Thermoregulatory Sweat Test If an indicator powder is used to demonstrate thermoregulatory sweating, a color digital camera can take images of the body at the end of the test. A paint program and a standard 444 Thermoregulatory Sweating studies and electromyography and clinical neurological examination are often normal in these patients, whereas tests of skin autonomic innervation are usually abnormal. This test compares favorably with and complements other tests of peripheral autonomic function, particularly in showing the whole body distribution of the neuropathic process. Hsieh C, McNeeley K, and Chelimsky T (2001) the clinical thermoregulatory sweat test induces maximal sweating. Introduction Thiamine is synthesized by many plants but only to a very limited extent by mammals. Intestinal microorganisms may be capable of synthesizing a small amount of thiamine in humans, but there are exogenous requirements.

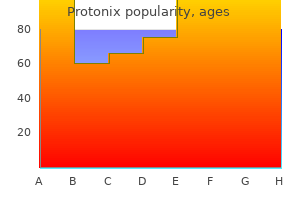
Generic protonix 20mg
Taking sequestrants with breakfast is desirable as it coincides with emptying of the gallbladder gastritis diarrhea order 40mg protonix fast delivery, which fills overnight, and facilitates maximum bile acid binding. Resins should be given with caution in patients with hypertriglyceridaemia as they can worsen this. Interruption of the enterohepatic recirculation of bile acids has important effects on hepatic lipoprotein metabolism. Sequestrants are not systemically absorbed so that other side effects are restricted to intestinal bloating and constipation. In consequence, they are very safe and can be used in pregnancy and during lactation. They are efficacious when given with statins or ezetimibe, but need to be taken several hours apart from other drugs and vitamins, which they bind to and prevent their absorption. They can be taken with statins and ezetimibe for the management of severe hypercholesterolaemia. They are also indicated in those with serious statin side effects, and in pregnancy and lactation. Nicotinic acid Nicotinic acid, also called niacin or vitamin B3, has been used to lower lipids over many years, particularly in the United States of America, but is no longer recommend for use in Europe (see next paragraph). Clinical trials, however, have shown that combination use of statins with drugs containing nicotinic acid did not lead to additional benefits in reducing the risk of major vascular events such as heart attack and stroke, but did result in a higher frequency of nonfatal but serious gastrointestinal events and infection. Nicotinic acid therapy is therefore usually started at a low dose and slowly increased to higher doses, under the cover of aspirin to reduce prostaglandin activity and flushing. Nicotinic acid can cause dyspepsia, mild increases in transaminases, and plasma uric acid. Their initial target is genetic hypercholesterolaemia, where cholesterol lowering is not adequate, or when there are severe statin side effects. Dietary supplements Supplements to the diet with plant sterols or stanols (such as FloraProActive or Benecol respectively) which compete for cholesterol absorption thus reducing plasma cholesterol levels can be used as an adjunct to lifestyle measures. Its side effects are mechanism of action based, with intestinal upsets due to fat malabsorption and fatty liver. Another drug is mipomersen a second-generation 2-Omethoxyethyl chimeric antisense oligonucleotide, which inhibits the synthesis of apoB. As with lomitapide, mipomersen causes intestinal upsets due to fat malabsorption and fatty liver. It has not been approved in Europe due to a 50 to 70% rate of side effects, mainly injection site reactions, flu-like symptoms, liver enzyme elevations, and proteinuria. Apheresis is performed weekly or twice-monthly depending on the degree of lipid lowering achieved. The main disadvantage is long-term access to the circulation, which is best achieved by an arteriovenous fistula, but venous access or a central line can be used. Further improvement is hoped for and anticipated with the drugs described previously described. It is a serious atherosclerotic cardiovascular disease risk factor and a risk factor for calcific aortic stenosis. Whether this applies with very high Lp(a) levels (>100 mg/dl (250 nmol/litre) is not known. In patients with high Lp(a) and symptomatic atherosclerotic cardiovascular disease, apheresis is effective in reducing Lp(a) levels and has potential to reduce disease progression, but its use is unlikely to be commonplace. A secondary goal is therefore to reduce the risk of atherosclerotic cardiovascular disease. Although in practice acute pancreatitis is rare with levels below 15 mmol/litre (1300 mg/dl), fat consumption can readily achieve this level in the predisposed. There are also patients, however, with persistent marked hypertriglyceridaemia who never develop pancreatitis. Pancreatitis is therefore an unpredictable complication of hypertriglyceridaemia and usually strikes unexpectedly. Lifestyle Moderately severe hypertriglyceridaemia (<15 mmol/litre (1300 mg/dl)) in the absence of chylomicrons (type 4 hyperlipidaemia) can be managed in the outpatient setting. A reasonable dietary goal is to restrict total fat intake to around 20 to 30 g daily. This is not always easy to achieve, because normal consumption is approximately 70 g daily, and requires dedication from the patient in understanding their dietary fat consumption. A formal dietary consultation and regular review with a dietitian with specific experience in the management of severe hypertriglyceridaemia is desirable. An extremely low-fat diet, less than 10 g of fat daily, for about 3 days is necessary (Table 12. This strict low-fat diet is not easy to maintain and not nutritionally adequate in the long term. Secondary factors Other factors contributing to hypertriglyceridaemia need to be actively sought and treated. In clinical practice, the most common problem is either undiagnosed or uncontrolled diabetes. In susceptible individuals, certain drugs, such as oestrogen, steroids, retinoids, and protease inhibitors, can also trigger hypertriglyceridaemia (Table 12. Further information on the secondary causes of hypertriglyceridaemia is found in Table 12. Fibrates and omega-3 fatty acids derived from fish are the only drugs available to treat hypertriglyceridaemia in the United Kingdom. They do not very much decrease atherosclerotic cardiovascular disease events due to hypercholesterolaemia, but are efficacious in hypertriglyceridaemia. They are safe and have few side effects, but can increase the likelihood of gallstones. Fibrates (particularly gemfibrozil) are associated with toxic myopathy especially when combined with statins or nicotinic acid (Table 12. Care and appropriate monitoring is needed in patients on anticoagulants and some diabetic blood glucose-reducing drugs as fibrates interact with these classes of drug. In the light of recent reanalysis of clinical trials of fibrates, their use in treating mild to moderate hypertriglyceridaemia and preventing atherosclerotic cardiovascular disease needs reconsideration. Omega-3 fatty acids Omega-3 polyunsaturated fatty acids or fish oils are present in high concentration in oily fish. They come from a variety of plants sources, but omega-3 fatty acids of plant origin are less well studied, and are not a recommended substitute for fish oils. Eicosapentaenoic acid and docosahexaenoic acid are the main active ingredients in fish oil. Fish oils are effective for the treatment of moderate hypertriglyceridaemia with levels of approximately 5 mmol/litre (450 mg/ dl). With more severe hypertriglyceridaemia, they are good in combination with fibrates. Higher doses of up to 12 g have been used with apparent safety and efficacy in very severe hypertriglyceridaemia, but should not be used in pure type 1 hyperlipidaemia, where they may exacerbate the phenotype. High doses can also be used with apparent safety in hypertriglyceridaemia during pregnancy. It is important that the fish oil is purified to remove mercury, dioxins, polychlorinated biphenyls, and other toxins that contaminate fish, particularly if prolonged use is anticipated or in pregnancy. The pathogenesis of hypertriglyceridaemic acute pancreatitis remains ill-understood. A likely precipitating factor is sludging of the very large chylomicron particles in the microvasculature of the pancreas leading to leakage of pancreatic enzymes into the circulation. Accurate measurement of serum amylase is challenging in the presence of lipidaemia and pancreatitis may be falsely ruled out when the amylase is not apparently elevated. In such situations, hypertriglyceridaemia may have improved markedly and may then be erroneously excluded as a possible cause of pancreatitis. The treatment of hypertriglyceridaemic pancreatitis does not differ greatly from that of pancreatitis of any other cause. Should total parenteral nutrition be necessary, it is important to avoid excess fat supply Other therapeutic measures in order to correct the hypertriglyceridemia include the use of low molecular weight heparin and insulin. In the early stages of recurrent acute pancreatitis and in pregnancy it may have value. There is, however, no evidence that patients treated with apheresis recover more rapidly or have fewer pancreatitis-associated complications, or have reduced mortality. Gestational diabetes may exacerbate this, and needs appropriate control if necessary with metformin and insulin. Doses of fish oils well above the usually recommended daily maximum of 4 g can be given with apparent efficacy and safety; up to 12 g appears efficacious.
Buy protonix 20mg with amex
The portion of the amygdala located in the uncus has abundant connections with olfactory structures gastritis cats discount protonix uk, such as the olfactory bulb and the peri- and entorhinal cortices. As the process worsens, the parahippocampal gyrus herniates as well, compressing the midbrain laterally. The midbrain then flattens anteroposteriorly, stretching its feeding arteries that run anteroposteriorly from the top of the basilar artery until they begin to tear. Subsequently, hemorrhages can appear in the tegmentum of the midbrain (Duret hemorrhages). In addition to ipsilateral third nerve palsy and hemiparesis, contralateral homonymous hemianopsia will result from compression of the ipsilateral posterior cerebral artery. If untreated, the brainstem distortion will ultimately result in decorticate posture, respiratory depression, and death. Mori K, Ishimaru S, and Maeda M (1998) Unco-parahippo-campectomy for direct surgical treatment of downward transtentorial herniation. This article is a revision of the previous edition article by Robert Daroff, volume 4, p 614, r 2003, Elsevier Inc. From there it extends into the cerebral peduncle of the midbrain, basis pontis of the pons, and pyramid of the medulla oblongata, decussating at the level of the inferior medulla to the contralateral corticospinal tract in the dorsolateral cervical spinal cord. Within the brainstem, corticobulbar fibers leave the tract to innervate cranial nerve motor nuclei. Coordinated movement is much more complicated than this, with the extrapyramidal, vestibular, and sensory systems interacting with these neurons, but this two-neuron system is the basic substrate for voluntary movement. Diaschisis can typically last hours to days following cerebral lesions, and sometimes weeks following spinal cord level lesions. Increased tone takes the form of spasticity, defined as velocity-dependent resistance to stretch. This is the result of a lack of inhibition with resultant increased contraction of involved muscles. In humans, the flexor muscles of the upper extremity and extensor muscles of the lower extremity are primarily involved as demonstrated by hemiparetic individuals with the lower extremity extended (which can be an aid to ambulation) and upper extremity flexed at the elbow and wrist, and often held across the abdomen. Persistent untreated spasticity can ultimately lead to contractures with resultant immobility of the involved joints. Spasticity needs to be distinguished from the rigidity seen in association with some extrapyramidal disorders. If the lesion results in partial disruption, the result is usually paresis rather than plegia of the limb(s). Distal limb muscles tend to be impacted to a greater extent than proximal limb muscles although this is not an ironclad rule. Thus, the familiar clinical presentation of 584 Encyclopedia of the Neurological Sciences, Volume 4 doi:10. This pattern is unlike that of spasticity, which, as stated above, primarily impacts flexor muscles of the upper extremity and extensor muscles of the lower extremity. Clonus Clonus tends to coexist with spasticity and, in a sense, is a manifestation of spasticity although it is not seen in all spastic patients. It is generally most easily demonstrated at the ankle, although it can be detected at the patella and occasionally in the upper extremity. It results from alternating and involuntary muscular contracture and relaxation in rapid succession. Clonus can also be spontaneous and generated by routine movements of an involved extremity. It commonly occurs when an individual retires to bed with associated voluntary stretching of the paretic spastic limb. Although the Babinski sign is the best known, there are a number of other ways to generate an upgoing toe that are manifestations of the activation of heightened withdrawal reflexes similar to the heightened deep tendon reflex. Associated problems often include aphasia, dysphagia, contralateral hemisensory disturbances, and contralateral visual field loss. In this setting, the absence of associated symptoms and signs helps to define the location of the lesion. Given the small diameter of the spinal cord, extraaxial, as well as intraaxial, lesions causing compression often impact the corticospinal tracts bilaterally. This can lead to quadriparesis with cervical cord lesions or paraparesis with thoracic cord lesions. The distribution of the sensory loss can be extremely helpful in localizing the level of a lesion. In this setting, an individual will have ipsilateral weakness and proprioception loss, along with contralateral loss of pain and temperature sense. Also, important is recognizing the occasional individual with psychogenic weakness in the form of a conversion reaction or malingering. In the case of hemiparesis, a careful history and physical examination can often lead to the correct diagnosis. The patient with psychogenic hemiparesis will have normal reflexes, no Babinski sign or clonus, and normal tone. The patients are then asked to press their heel downward, as strongly as possible. In doing so, patients with psychogenic weakness exert stronger downward pressure with the opposite heel than they did when asked to do so voluntarily, thus indicating that the weakness is not organic. Definition Uremia is a condition caused by renal failure and excessive accumulation of products of protein metabolism, as well as loss of intrinsic kidney homeostatic and endocrine function. Phenytoin, carbamazepine, and valproate, which are not cleared by the kidney, are suitable agents. Caution needs to be used with renally excreted antiepileptic drugs, for example, levetiracetam and gabapentin. Pathogenesis of Uremic Encephalopathy Clinical Manifestations of Acute and Chronic Uremic Encephalopathy Uremic encephalopathy can be divided into acute and chronic varieties, although there are considerable overlaps. Some of the most likely chemicals include guanidino compounds, parathyroid hormone, urea, and various middle molecules. Encephalopathy Related to Treatment of Uremia Acute Uremic Encephalopathy Acute uremic encephalopathy is a florid illness ranging from subtle executive dysfunction to coma. Delirium, either hypoactive or hyperactive, is the first stage with fluctuating, reduced attention, impaired construction and writing, executive dysfunction, behavioral changes, and sleep disturbances. Hyperventilation, with Kussmaul breathing, occurs during periods of metabolic acidosis. Motor findings include paratonia, multifocal myoclonus, action myoclonus, stimulussensitive myoclonus, tremor, asterixis, and seizures. Uremic coma, now uncommon, is typically accompanied by Kussmaul breathing related to metabolic acidosis. Dialysis Disequilibrium Dialysis disequilibrium, affecting patients during or shortly after hemodialysis, is caused by shifts of water, causing cerebral edema. Headache, nausea, vomiting, restlessness, myoclonus, disorientation, and somnolence are the main features. More severe manifestations include organic psychosis, generalized seizures, stupor, or coma. In addition, the anorexia, nausea, and vomiting, present in some patients with chronic uremia, may lead to dramatic reduction in oral thiamine intake. As in acute uremic encephalopathy, postural or kinetic tremor, myoclonus, and asterixis can occur. Although generalized convulsive seizures occur in chronic uremic encephalopathy, they are typically seen at the very end stage of the disease, and may be accompanied by stupor or coma. With improved treatment strategies, Encyclopedia of the Neurological Sciences, Volume 4 doi:10. Furthermore, microhemorrhages and infarcts are occasionally seen in addition to infarction. Finally, cortical gray matter, in addition to underlying white matter, can be involved. In general, therapy is targeted at managing hypertension, withdrawal of the offending agent, and treating the underlying condition. Although less common with aggressive dialysis, frontally predominant triphasic waves with a frontooccipital gradient can be seen in severe azotemia or in decompensated chronic renal failure. Management Most of the features of uremic encephalopathy and uremic neuropathy do not completely resolve with dialytic therapy.
Buy protonix line
From physiological studies xango gastritis order protonix 20mg without a prescription, the startle response is a pathological exaggeration of the normal startle reflex. In hereditary or primary forms of startle disorders, abnormal startle responses occur as isolated problems without other neurological or medical signs, whereas in secondary startle syndromes the heightened response to startle occurs as an associated problem within the context of a specific illness. Hereditary hyperekplexia is an autosomally dominant disorder related to mutation in the a1 subunit of the inhibitory glycine receptor protein localized on chromosome 5q33. Recently, recessive and compound hetrozygote modes of transmission have also been described. The hallmark of hyperekplexia is an exaggerated startle response to a sudden, unexpected stimulus. Typically, this startle response extends to the lower extremities and is resistant to habituation. Newborns with hyperekplexia demonstrate a continual stiffness even when left quietly, and superimposed on this hypertonicity, dramatic flexor postures develop whenever noise startles them or they are touched. Unlike the normal baby reflex called the Moro response, the child reacts to noise by flexing the arms rather than the expected extension. Cessation of breathing and cardiorespiratory arrest can occur, perhaps from stiffness of the chest wall. As the child matures over several months, the constant increased tone gradually disappears but the abnormal startle response persists and is typically followed by delayed generalized stiffening that may interfere with walking and eating, resulting in falls and choking. Severely affected patients have startle attacks throughout life, and this often worsens during adolescence with variable improvement later in life. Although adults may have only infrequent bouts of heightened startle responses, stress and superimposed medical illnesses generally precipitate a new series of episodes. Violent bilateral flexion of the legs can occur as patients fall into slow-wave sleep, causing them to injure themselves or their bed partner. Abdominal muscle involvement has been postulated to occasionally result in inguinal and abdominal hernias. There have been a few unusual clusters of subjects with abnormal startle responses that are of particular historical interest and poorly classified. Although abnormal startle responses occur in these subjects, additional behavioral components are likely to be culturally based, conditioned behaviors. As a group, subjects demonstrate an initial violent startle in response to a sudden stimulus that is variably followed or accompanied by automatic, repetitive, and sometimes socially unacceptable speech, repetitive imitative gesturing, and peculiar postures. The startle reflex in hyperekplexia is believed to be mediated by brainstem rather than cortical mechanisms. This mechanism is supported by electromyographic evidence of a caudorostral direction of recruitment of cranial muscles during startles induced by tapping the forehead, and by lack of electroencephalographic correlates. Further support comes from the observation that sporadic hyperekplexia is commonly caused by structural lesions in the brainstem, particularly pontine reticular nuclei that regulate physiological auditory startle reflex. Clinically, hyperekplexia should be differentiated from reticular reflex myoclonus, startle epilepsy, and startle-induced tics. Hyperekplexia is distinguished by stimulus sensitivity in the mantle area and the presence of tonic stiffening spasms, whereas in reticular reflex myoclonus the stimulus sensitivity involves the limbs, the spread of movements is faster, and there are spontaneous jerks in addition to induced jerks. Clonazepam and other benzodiazepines are considered the drugs of choice for hyperekplexia, and treatment reduces the severity and frequency of the startle reactions. Valproic acid, 5-hydroxytryptophan, and piracetam have also been successful in certain patients. It usually presents as repeated brief, time-limited convulsions, without recovery to full alertness and normal mental function between seizures. Extensive clinical descriptions were provided in the late nineteenth and early twentieth centuries by Bourneville, Trousseau, and Clark and Prout. International conferences were subsequently held in 1980, 1997, 2007, 2009, and 2011. This suggests a likely worldwide incidence of approximately 3 million per year, but adequate epidemiological data are not available to determine the worldwide incidence accurately determine the worldwide incidence. The clinical features of the motor or sensory seizure activity are dependent on the area of the cortex from which the seizures start. Over time, the ictal discharges begin to merge together to produce a waxing and waning pattern, which is followed by continuous, generally monomorphic, ictal discharges that may persist for some time. There is controversy regarding the interpretation of periodic epileptiform discharges, and some authors have considered these injury patterns, rather than an expression of ongoing seizure activity. Draw blood for serum chemistry, hematological values, and antiepileptic drug concentrations. Patients have recovered consciousness and useful existence after 42 months of coma. Status Epilepticus 299 seizure-stopping mechanisms, or the occurrence of a strong excitatory stimulus, may result in repeated or prolonged seizures. A variety of acute neurological insults either lower seizure threshold or result in excessive excitation or inhibitory failure. A recent study demonstrated the utility of prehospital emergency treatment by paramedics using intravenous diazepam or lorazepam, and another recent prehospital treatment study found intramuscular midazolam to be at least as effective as intravenous lorazepam. Lorazepam is the most common benzodiazepine used, although diazepam, midazolam, or clonazepam (widely used in Europe) is also sometimes the initial drug. Intravenous general anesthesia is induced by continuous infusion of pentobarbital, midazolam, propofol, or sometimes diazepam or lorazepam. Shorvon S (1994) Status Epilepticus: Its Clinical Features and Treatment in Children and Adults. Biology of Stem Cells the discovery that a fertilized egg can generate an entire organism as complex as a human being has fascinated scientists since the nineteenth century. Studies to understand this multifaceted process have revealed that with increased numbers of cell divisions, the initially totipotent cell. A transient population of embryonic stem cells formed early after fertilization are called pluripotent, having the potential to give rise to any cell type in the body. On undergoing several divisions, they become more specialized but are still able to generate various cell types within a lineage (multipotent) and eventually entire tissues and organs are formed. Although embryonic stem cells disappear during development, their multipotent progeny are able to self-renew and thus persist in many tissues throughout life. In the adult, the proliferative activity of these stem cells varies a lot depending on the tissue, but they often have vital functions in tissue regeneration and maintenance. These neuroepithelial cells transform into radial glial progenitor cells with apical-basal polarity that either directly generate neurons or give rise to intermediate progenitor cells, which in turn generate neurons and oligodendrocytes. Astrocytes are formed by transformation of radial glial cells after embryonic brain development. The reasons for these restrictions, at least under physiological conditions, are not entirely clear. Given that the integration of new neurons into an existing and well-balanced neuronal network might bear the danger of disturbance and malfunction, it is possible that there is an active suppression of adult neurogenesis in many brain regions. This could provide an explanation why cells isolated from other adult brain regions, such as cortex or cerebellum, can have stem cell potential once isolated and provided with the right mixture of mitogens and nutrients, although they are not generating newborn neurons under physiological conditions in vivo. Furthermore, adult neurogenesis does not occur in such an orchestrated manner as during development. Although cell proliferation and neuronal differentiation occur wave-like in the developing brain, various stages, from proliferating cells to cells that need to establish dendritic connections are present in close proximity at the same time in the adult brain. Thus, newborn cells are likely to have distinct individual needs that might only be supported in the two neurogenic areas of the adult mammalian brain. Stem Cells in the Adult Brain As mentioned earlier, multipotent stem cells can self-renew and persist throughout life and are, for instance, constantly renewing our skin, gut, and blood. In the brain, however, it was for a long time believed that no new neurons are formed after development is completed. The initial findings in the 1960s by Altman and colleagues that there are dividing cells throughout life in the mammalian brain were not perceived without skepticism. It took several decades and many more experiments until the existence of stem cells that can proliferate and generate up to three neural lineages in the adult brain became generally accepted. Neuronal Differentiation and Activity-Dependent Integration the complex process of differentiation and integration of adultborn neurons has been the topic of many studies. Labeling techniques such as retrovirus-mediated expression of fluorescent proteins specifically in newborn neurons or injection of modified thymidine analogs that become integrated into the deoxyribonucleic acid of dividing cells and can be used for birth dating together with immunohistological makers for different maturation stages, made the developmental process of neuronal differentiation more accessible to be studied in vivo. As during embryonic development, there is also an excess production of newborn neurons in the adult, the majority of which dies during the maturation and integration process. Those newborn neurons that finally get integrated go through well-described distinct stages, similar to the ones during embryonic and postnatal development but over a more extended time.

Purchase protonix with mastercard
However gastritis and diet pills discount 40 mg protonix amex, the technique can be difficult, and caution must be exercised not to puncture the pleura and cause pneumothorax. Myopathies (including facioscapulohumeral muscular dystrophy) usually cause bilateral winging, which can be asymmetrical. Secondary scapular winging also occurs when pathology of the glenohumeral joint interrupts the coordinated motion of the scapula. Such false-negative diagnoses include acromioclavicular joint injury, posterior glenohumeral joint instability, rotator cuff tear, and aseptic necrosis of the humeral head. Acute transections are best managed with repair and nerve grafting, when necessary. Such conservative treatment is commonly used with idiopathic brachial plexus neuropathy, although recovery may take many months to several years. More than half of these patients cannot lift or pull heavy objects, participate in sports, or work with hands above shoulder level. Initial treatment consists of activity modification, analgesics, and nonsteroidal anti-inflammatory drugs. Physical therapy should include the range of motion exercise and stretching and strengthening of the scapular stabilizers, rotator cuff, and cervical muscles. The interscalene triangle is bordered by the anterior scalene muscle anteriorly, middle scalene posteriorly, and medial surface of the first rib. With the latter, tissue destruction may be substantial enough to require amputation. The diagnosis is confirmed by the demonstration of reduced arterial blood flow in the limb, usually by noninvasive followed by invasive vascular procedures. Nonetheless, they are due to ischemia of the distal nerves and not to ischemia of the brachial plexus. It is a rare, unilateral disorder that characteristically affects adults, often young adults, without a specific sex preference. Distal to the narrowed arterial lumen, the blood flow becomes turbulent, resulting in aneurysm formation. Thrombi form on the aneurysmal wall, and from them emboli arise and pass downstream, where they may occlude smaller vessels, thereby causing ischemia. From the tip of either of Encyclopedia of the Neurological Sciences, Volume 4 doi:10. Either the distal portion of the T1 anterior primary ramus (before where it fuses with the C8 anterior primary ramus to form the lower trunk) or the proximal lower trunk is stretched and angulated around this band. A predominantly motor syndrome results, consisting of weakness and wasting of the hand and often some of the forearm muscles. Frequently, the hand wasting (but not the weakness) is selective, restricted to the lateral thenar. Although the majority of patients subsequently admit to having experienced intermittent aching for many years along the medial arm, forearm, and sometimes medial two digits, typically these sensory complaints were never severe enough to cause them to seek medical attention or even to mention them to physicians. Neurological examination reveals the hand wasting, characteristically accompanied by global hand weakness. Some surgeons prefer to remove the bony anomaly, or at least a portion of it, as well. Many patients report experiencing some improvement in hand strength soon after surgery, but this is never substantial and is never accompanied by reversal of the hand wasting. If hand cramping with use was present before surgery, it usually persists, although often in a less severe form. In contrast, the sensory symptoms experienced in a T1 distribution are usually relieved, and the hand weakness and wasting are permanently arrested. A pertinent point is that the affected hand almost never regains its normal bulk or strength, because this never occurs when a very severe and very chronic axon-loss lower trunk brachial plexopathy is treated, regardless of the cause. It is essentially an offspring of the discredited scalenus anticus syndrome of an earlier era. Although a few proponents contend that both arterial and neural structures are compromised, most believe that only portions of the brachial plexus are involved. The most commonly invoked cause has been trauma, either of the acute, single-episode variety (especially whiplash-type injuries following rear-end vehicular accidents) or due to repetitive limb use, such as is characteristic of factory and office workers. Unfortunately, there is no unanimity of opinion regarding which test is most helpful in this regard. Probably the majority of proponents use the 3-min elevated arm stress test (maneuver) for this purpose. It is not surprising that, for a condition of doubtful existence, many physicians believe that it is overdiagnosed. Many of the skeptics are neurologists, whereas many of the proponents are surgeons, particularly thoracic or vascular surgeons. Unfortunately, the majority of earlier reports on this subject were provided by the operating surgeons after brief follow-up periods. These reported results illustrate the importance of bias being removed from surgical trials. Recently, with longer follow-up periods and postoperative assessments performed by researchers other than surgical teams, the results have been far less impressive. It is usually due to some focal clavicular abnormality, most often a midshaft fracture. The proximal portion of the axillary artery and vein and the cords of the brachial plexus can be injured as a result of this fracture, either alone or in combination. Symptoms and signs are characteristically present both locally, at the fracture site, and in the affected limb. Local findings may include focal tenderness, a bony deformity, a mass beneath the damaged portion of the clavicle, or a bruit. The limb changes depend on the particular neurovascular structure(s) injured but may include diffuse limb swelling, loss of distal pulses, pain, paresthesias, weakness, and loss of deep tendon reflexes. Many patients require surgery, both on the clavicle and on the structures beneath it that have been damaged. Unfortunately, the most commonly damaged brachial plexus element is the medial cord, and such injuries generally cause permanent residuals. The confusion is a direct result of five very distinct entities being included under the same name. Thoracic Nerve, Long Further Reading Cherington M (1989) A conservative point of view of the thoracic outlet syndrome. Cherington M, Happer I, Mechanic B, and Parry L (1986) Surgery for thoracic outlet syndrome may be hazardous to your health. Thoracic Spine Stabilization J Kalyvas and N Theodore, Barrow Neurological Institute, St. Introduction Spine stabilization refers to the surgical procedures that correct spinal instability and deformity while they optimize the physiological weight-bearing function and mobility of the spinal column. With a variety of rigid hardware constructs tailored to specific spinal pathology, surgical approaches, and goals, internal fixation allows bony fusion (arthrodesis) to occur across vertebral segments for definitive long-term stabilization. Stabilization of the thoracic spine offers unique surgical advantages and challenges related to its relationship with the rib cage and surrounding vital structures, which warrant special consideration. This article addresses these issues as well as the surgical indications, approaches, techniques, and appropriate pre- and postoperative management related to stabilization of the thoracic spine. Consequently, anterior column fractures and kyphotic deformities are the most common. Compression Fractures Compression fractures involve collapse of the anterior column, which results from axial loading and a flexion moment. By definition, these fractures do not involve the middle column or threaten the spinal canal. Burst Fractures Surgical Indication Spinal instability requiring surgical fixation can be defined as structural changes under physiological loads, which alter the relationship between the vertebrae. The instability results from a wide variety of pathologies that affect other regions of the spine, including senescent degeneration, neoplasms, infections, trauma, or instability from previous surgical intervention. When two of the three columns are disrupted, instability is implied and surgical fixation is indicated. Fractures of the thoracic spine are the most common cause of instability, and they follow characteristic patterns because of the unique biomechanics of this region. Specifically, the middle and posterior columns of the first nine thoracic vertebrae have greater stability during extension compared with the rest of the spine because of their articulations with the rib cage and sternum. The anterior column, however, is particularly susceptible to vertical forces during flexion Burst fractures involve collapse of the anterior and middle columns, which results from axial loading and a flexion moment.

Buy protonix 20mg overnight delivery
Porphobilinogen deaminase (hydroxymethylbilane synthase) Bialleic mutations in the third enzyme in the haem biosynthetic pathway led gastritis diet underactive thyroid cheap protonix online visa, in one well-studied pedigree, to an unusual neurological syndrome with impaired cerebellar function and slowly progressive white matter disease with hypomyelination. There was also truncal muscle weakness and wasting, notably associated with ptosis, a clinical sign compatible with mitochondrial disease. Biochemical tests revealed excessive urinary porphobilinogen, 5-aminolaevulinate, porphobilinogen, and uroporphyrin. Furthermore hepta-, hexa-, penta-, and coproporphyrins I were highly increased in urine and typical of patients with acute porphyria during a metabolic crisis. The author suggests that these features indeed indicate a mitochondrial defect, probably due to impaired oxidative phosphorylation related to the haem deficiency in critical cytochrome components of the electron transport chain, such as cytochrome c. Supportive evidence is provided by defective mitochondrial energetics in the brain and skeletal muscle of mice generated as homozygous mutants for the porphobilinogen deaminase locus, and by a patient having consistently elevated plasma lactate concentrations and metabolic acidosis. Intermittent hypoglycaemia was found in one patient who died at the age of 20 years due to cardiorespiratory failure; their sibling had died suddenly at 9 years of age of unknown causes. Coproporphyrinogen oxidase this is a very rare syndrome principally dominated by haematological (erythroid) features of haemolytic anaemia accompanied by hepatosplenomegaly and marked photosensitivity. Most patients have a mild to severe haemolysis with increased reticulocytosis, circulating normoblasts, decreased serum haptoglobin, and increased unconjugated bilirubin concentrations. Inclusion bodies are often seen in marrow, erythroid cells, and circulating normoblasts. Splenomegaly develops in childhood, thereby causing pancytopenia as a result of hypersplenism; this accelerates the haemolysis and leads to compensatory erythropoiesis in the bone marrow. The classic skin manifestations are of severe blistering lesions on sunlight-exposed skin, particularly of the hands and face, with the 12. Healing of the lesions with or without consequential infection often leads to cutaneous deformities with loss of digits, scarring of the eyelids, nose, lips, ears, and scalp, and occasionally blindness due to corneal scarring. Examination of the teeth shows erythrodontia and deformities, and exposure to ultraviolet light may reveal striking dental fluorescence. These reduced and colourless metabolites become oxidized to the fluorescent tissue and urinary porphyrins associated with the passage of pink urine that characterizes this often devastating disease. Splenectomy is often required in early childhood to reduce transfusion requirements and improve cytopenias. This may lead to an innovative treatment for this severe and destructive disease, which currently lacks molecular therapy. Porphyria cutanea tarda this disease is the most common of the cutaneous porphyrias and, unlike other hepatic porphyrias, is never associated with acute porphyric crises. Aetiology Toxic cutaneous porphyria may result from environmental exposure to dioxin or to hexachlorobenzene, particularly after industrial accidents such as that which occurred in Turkey in the 1960s. Occasional cases have been reported after exposure to other halogenated phenols, but under these circumstances it appears simply to be an environmental toxic syndrome which is separate from the sporadic porphyria cutanea tarda that is precipitated by other specific environmental factors including increased hepatic storage iron, excess ethanol consumption, administration of oestrogens, hepatitis C virus infection, human immunodeficiency virus infection, and (possibly) nutritional deficiencies including antioxidants such as vitamin C. There is a clear association with renal impairment in which the development of the disease can be explained by the presence of iron overload (as a result of defective iron utilization with or without routine iron supplementation, particularly in patients on haemodialysis) and failure to excrete excess plasma porphyrins that do not readily diffuse through the peritoneal cavity or haemodialysis membranes. The sequencing of the human uroporphyrinogen decarboxylase gene that maps to human chromosome 1p34 has not provided any evidence of mutations to account for the tissue-specific enzyme deficiency, and no isoforms of the enzyme have yet been identified, hence the molecular pathogenesis of sporadic porphyria cutanea tarda remains unknown, but it is clear that iron and other environmental influences inactivate the hepatic enzyme. Less than one-quarter of patients who have porphyria cutanea tarda show a familial susceptibility to the condition, when mutations in one allele of the human uroporphyrinogen decarboxylase gene lead to catalytic deficiency of the enzyme in all cells, including erythrocytes. In most instances, the genetic defect leads to partial reduction of the enzyme protein encoded by the mutant allele. Studies of pedigrees affected by familial porphyria cutanea tarda indicate that expressivity of the trait is very low; less than 10% of heterozygotes develop clinical disease. Conversely, a very few patients present with a syndrome that closely resembles congenital erythropoietic porphyria with marked blistering skin lesions, excess hair growth, and cutaneous scarring in association with the excretion of pink or red urine. In hepatoerythropoietic porphyria, the activity of uroporphyrinogen decarboxylase is markedly deficient, although residual activity remains to preserve essential haem biosynthesis in the erythron and liver. Most patients ultimately develop splenomegaly with accelerated haemolysis closely resembling congenital erythropoietic porphyria. Molecular analysis of the human uroporphyrinogen decarboxylase gene may assist the prenatal diagnosis of at-risk pregnancies in women who have already given birth to an affected infant. Clinical features the clinical features of porphyria cutanea tarda of whatever form are very characteristic and are confined to light-exposed skin. Usually, the only signs are of erosions resulting from minor trauma in skin, with increased fragility as a result of light exposure, typically on the dorsum of the hands. Other changes include the development of large subepidermal bullae after exposure to light, which may burst leaving ulcerated lesions that are slow to heal. Increased pigmentation, often accompanied by areas of decreased pigmentation, is a common feature combined with increased hair growth, particularly on the face. Patients with porphyria cutanea tarda do not always notice the photosensitivity and rarely experience marked pain unless exposed to brilliant sunlight. Occasionally, there is evidence of dermal injury and loss of nails, damage to the conjunctivae, and hair loss. Careful examination of the affected areas shows small depigmented cutaneous scars and the formation of milia. If bacterial infection occurs and there is repeated exposure to sunlight, then severe and permanent scarring may result. Typically, porphyria cutanea tarda occurs in middle-aged men with a history of alcohol use and in women after institution of oestrogen replacement therapy; in young persons, infection with hepatitis C or the immunodeficiency virus may precipitate disease expression. Frank signs of hepatomegaly or iron overload are rare in porphyria cutanea tarda but have been noted; as with adult haemochromatosis, there is a significantly increased frequency of hepatocellular carcinoma. Occasionally patients with porphyria cutanea tarda may notice an increase in urine excretion of formed porphyrins which, especially after concentration overnight, may resemble the colour of tea or cola. The stool and urine contain large quantities of coproporphyrins and uroporphyrins that fluoresce intensely on exposure to longwavelength ultraviolet light when placed in a suitable vessel for its transmission (namely silica rather than standard glass). Similarly, examination of liver biopsy specimens under ultraviolet light reveals bright red/orange fluorescence; microscopic examination may also show coincidental hepatitis with or without excess deposits of stainable tissue iron reflecting the increased iron storage of this disease. Treatment Sunlight exposure should be avoided as much as possible and sunblock creams used until the porphyrin abnormality is corrected. Patients should be advised to moderate or stop their intake of alcohol and avoid the use of iron tonics and sex hormones, especially oestrogens. Screening should be undertaken for chronic infection with human immunodeficiency virus and hepatitis viruses, especially hepatitis C. Management should also include imaging or biopsy of the liver if serum liver-related tests are abnormal, as well as measurement of serum -fetoprotein since there is a risk of hepatocellular carcinoma in this disease. Most patients with porphyria cutanea tarda respond to iron depletion by phlebotomy and initial iron status should be determined by measuring serum ferritin concentrations. Weekly or fortnightly removal of 500 ml of blood will usually correct the abnormal urine and plasma porphyrin profile within a few months, but maintenance phlebotomy will be required, usually amounting to the removal of 2 to 4 units of blood at intervals each year. This man, a taxi driver, had noticed irritation after exposure of his hands to light transmitted through the windscreen. He had noticed fragility and blistering combined with pigmentary changes typical of this disorder. The cutaneous manifestations of porphyria cutanea tarda respond rapidly to low-dose chloroquine treatment, which should be considered in patients with persistent symptoms or at the outset before iron storage has been fully corrected. This action of chloroquine was discovered empirically, but the agent forms complexes with uroporphyrin deposits and promotes their external cellular disposal, promoting excretion of uroporphyrin from the liver and inducing marked but transient porphyrinuria. Although chloroquine usually provides rapid relief from the cutaneous disease and photosensitivity, it does not correct the underlying metabolic defect in the liver and its long-term use is not recommended unless the other provocative factors in porphyria cutanea tarda have been removed. The usual effective dose of chloroquine is 100 to 200mg given once or twice weekly; larger doses are associated with marked hepatic toxicity in porphyria cutanea tarda. The drug is reported to have no therapeutic effect on other photosensitive porphyrias. These findings are reflected in a large series of 226 patients with a clinical and biochemical diagnosis of protoporphyria from the United States of America. A ferrochelatase mutation and the common low-expression mutation was detected (presumed in trans) in 186 (82. These are early findings and more comprehensive genetic studies are needed fully to explore the role of ClpX mutations in erythropoietic protoporphyria. Females with the trait may be asymptomatic and-as somatic mosaics-show greater variability of clinical expression than affected males within their pedigree.

40mg protonix mastercard
Attacks usually have a frequency of above five a day treating gastritis through diet discount protonix online master card, and respond completely to indomethacin. Since the initial description, more than 100 cases have described in the literature. Initially, the reported female: male ratio was 7:1, but in a subsequent review of 84 patients the female: male ratio was 2. The recent case series of 31 patients has not shown a female preponderance (M:F ratioB1:1). The physiology of this system is relatively well described and a degree of cranial autonomic activation is a normal response to a cranial nociceptive input. Whether this latter area holds the key to understanding the indomethacin effect in these syndromes remains one of the important questions for the next few years. Clinical Features Typically, the pain is in the ophthalmic division of the trigeminal nerve, but it can widely occur in the head. Lacrimation, nasal congestion, conjunctival injection, and rhinorrhoea are the most frequent symptoms and signs accompanying the attack. Attacks occur several times per day, with a mean of 11 and median duration of 19 min in one large case series. Family History and Genetics Migraine is typically described as a genetic headache disorder and mutations have been discovered in patients with familial hemiplegic migraine. Alcohol intake triggers pain in approximately onefifth of patients, whereas menstruation does not seem to be an important and consistent trigger and the possible role of pregnancy has not been documented. This release is likely to mark trigeminovascular and cranial parasympathetic 508 Encyclopedia of the Neurological Sciences, Volume 4 doi:10. In the episodic form attacks occur in periods lasting from 7 days to 1 year, separated by free periods lasting 1 month or longer. In the chronic form, attacks occur for more than 1 year without remission or with remission lasting less than 1 month. However, longer attacks can occur, with 600 s of the so-called saw-tooth pattern having been recorded in the largest case series reported. Differential Diagnosis of Paroxysmal Hemicrania Secondary Paroxysmal Hemicrania Probably as a result of publication bias, there are a number of case reports of symptomatic cases in the literature, though a causal relationship with the underlying structural lesion is uncertain in many cases. Second, an increased intrasellar pressure may play a role in the genesis of the pain. Finally, it has been suggested that the pathogenesis of the attacks is mainly neurohormonally mediated rather than by the size or invasiveness of the tumor. A volume effect seems unlikely as the presentation and disability associated with pituitary tumor-related headaches is unrelated to their size. The attacks are never accompanied by prominent conjunctival injection, although slight lacrimation may infrequently be noted. Both conditions are strictly unilateral, relatively brief, frequent headaches that occur in association with ipsilateral cranial autonomic features. At present, the absolute response to indomethacin is the only crucial factor that permits a distinction between these two conditions. The available evidence suggests that it is a lifelong condition with a mean duration of illness of 13. Indomethacin does not seem to alter the condition in the long term, though a significant proportion of patients can decrease the dose of indomethacin required to maintain a pain-free state over time. The indomethacin test can be performed in two different ways: the oral trial or a placebo-controlled indotest test. Daily dosing of indomethacin for a long period may be troublesome mainly due to gastrointestinal adverse effects. The parenteral trial consists of singleblind administration of 100 or 200 mg, or both and placebo. It has been suggested that placebo-controlled may be the gold standard, when applied. However, the prolonged use of these agents has been linked to an increased risk of myocardial infarction and strokes that led to the worldwide withdrawal of rofecoxib from the market. Topiramate and verapamil have both been reported to be useful in several case reports. With longer attacks, both response and lack of response to sumatriptan have been reported. Greater occipital nerve block with lidocaine and methylprednisolone has a positive effect in some patients. This is in contrast to cluster headache, where the majority of cases are of the episodic form. The pain is usually around the eye and in the retroorbital region, but can be anywhere in the face or on the ipsilateral side of the head. Attacks can be spontaneous, or triggered by touching the side of the face, chewing, talking, cold wind on the face, and brushing the teeth, amongst others. The International Headache Society Classification Criteria for Pain (verbal rating scale from 0 to 10) 1. The purported mechanism in posttraumatic migraine is axonal injury or a shearing effect to the brainstem even after mild or moderate injury, which would cause a physiological shift in the brainstem, leading to chronic migraine. However, a small but significant proportion of patients have an associated abnormality, usually in the posterior fossa or the brainstem. There is also a group of patients with pituitary adenomata, either micro- or macroprolactinoma, with headaches secondary to these. During an attack the cranial autonomic signs may be observed, but these abate quickly at the end of the attack. However, there is a minority of patients with abnormal interictal findings on examination, such as reduction of sensation or hyperesthesia to pinprick. A personal or family history of headache including migraine is also useful, as is a list of previous medications and response to them, including indomethacin and inhaled oxygen. Short-Term Preventive Medication Short-term prevention in the form of intravenous lidocaine (1. Therefore, the patient must have cardiac monitoring for the duration of the infusion, and the rate of infusion should be slowed if psychological adverse effects develop. Lidocaine can be given for up to 7 days, which may allow the patient at least some short-term relief from pain. Freedom from attacks has been reported up to 6 months after an infusion, but any length of remission may allow upward dose titration of a preventive medication. Corticosteroids Steroid treatment is used in the short-term preventive treatment of cluster headache, where it can allow upward titration of longer-use preventive medication. However, as previously described, treatment for pituitary adenomata, either with dopamine agonists or hypophysectomy, can either ameliorate or worsen the attacks. The mechanism is a neuromodulatory process affecting the trigeminocervical complex, thus affording relief of symptoms for weeks up to a few months, and therefore allowing the titration of preventive medications. Lamotrigine In open-label studies, lamotrigine given in doses up to 300 mg daily has been reported as highly efficacious in a number of series. Patients with a previous history of renal stones, glaucoma, or depression, and those who are underweight should not be offered topiramate as a first-line agent, in order to prevent these known side effects. Another adverse effect of topiramate is cognitive slowing, which can be minimized by starting at a low dose (12. Therefore, trigeminal microvascular decompression may be considered in patients with arterial loops impinging on the trigeminal nerve. Side effects are limited to local adverse effects of the surgery and implantation of the stimulator, although one patient subsequently developed hemicrania continua, which was treated with indomethacin. Although the region of the posterior hypothalamus was targeted, more careful anatomical study has localized the target to the midbrain tegmentum. Matharu M and May A (2008) Functional and structural neuroimaging in trigeminal autonomic cephalalgias. Trigeminal Nerve (Cranial Nerve V) the trigeminal nerve or cranial nerve V is a mixed nerve that has three major branches: the ophthalmic division (V1), maxillary division (V2), and mandibular division (V3). The mandibular division is the only one to contain motor fibers responsible for controlling the muscles of mastication and other smaller muscles. The mesencephalic nucleus located in the lateral edge of the periaqueductal gray matter of the midbrain receives propioceptive information from the muscles of mastication and the other smaller muscles supplied by the trigeminal nerve.
Purchase 40mg protonix amex
Reports in the literature have shown that antibodies generally considered unimportant gastritis diet alkaline buy 40 mg protonix overnight delivery, can, under certain circumstances, be rendered significant and vice-versa. Closely matched blood can prevent immunisation to important antigens and lessens the subsequent burden of supplying compatible blood for patients who develop multiple antibodies. Not all workers agree with this approach, however, as not all patients will be responders to antigenic stimuli. The benefits must therefore be weighed against the cost of phenotyping both patients and donors. As a final note, blood should only be transfused when it will benefit the patient. Some applications of this technology and the methods applied are described in this chapter. Both of these procedures are invasive and associated with increased risks of spontaneous abortion. False predictions caused by these variants can be reduced by taking them into account in the design of the test. Trials of highthroughput methods have demonstrated that accurate fetal D testing in all D-negative pregnant women is feasible and it is now standard practice in Denmark and the Netherlands, avoiding unnecessary treatment of pregnant women with blood products. Complications can arise from null phenotypes, which usually result from inactivating mutations within the gene, meaning that no antigen is produced despite apparently being encoded. These null phenotypes are usually very rare, but they should be taken into account in any populations where they reach a significant frequency. Transfusion-dependent patients, such as those with haemoglobinopathies, often benefit from determination of their blood group phenotypes, either to assist in identification of their blood group antibodies or to provide matched blood so that they do not make multiple antibodies. This usually applies to patients with autoimmune haemolytic anaemia as a full predicted phenotype provides clues to which clinically significant alloantibodies might be masked by the presence of an autoantibody. Molecular tests can be used for testing patients and donors when serological reagents are of poor quality or in short supply. For example, antiDoa and -Dob can be haemolytic, yet satisfactory serological reagents for antigen testing are not available. Some Rh variants are relatively common in people of African origin, but are difficult to define serologically. Molecular tests can be employed to assist in finding suitable blood for sickle cell disease patients. The numerous variants of D result in reduced expression of D and loss of D epitopes (Chapter 4). In many cases D variants cannot be distinguished by serological methods, so molecular methods are required for their identification. Such a database would be very useful for providing blood for transfusion-dependent patients, such as sickle cell disease patients or patients with other haemoglobinopathies. Molecular methods are already starting to replace serological methods for some blood group typing, and this trend will continue. This is an extremely powerful technology that provides the capacity to sequence the whole genome of 10 people in one run, but also provides the capacity to sequence limited regions of the genome of many different individuals in one run. So it has the potential for testing all required blood group polymorphisms for numerous donors, in one session of sequencing. Probably antibody screening and identification will still require serological methods, though much of that will be done with synthetic, recombinant antigens. In transfusion, this is of prime importance and the ideal to be achieved is zero defects throughout the transfusion chain. In this way, the whole transfusion process can be analysed and improved in the long term. Single tests, for example, in an emergency situation, require that controls are included at the same time. A sample no older than 72 hours may be used if the last transfusion was between 14 and 28 days previously. Blood Administration Blood return Laboratory errors, root cause analysis (rCa), and corrective and preventive action (Capa) the purpose of pre-transfusion testing is to ensure that the right component of blood is selected and administered to the right patient. This involves many laboratory protocols where errors can and, unfortunately, do occur. Errors can occur at any time from the point of the tests being ordered to the final reporting of the results and therefore include errors of interpretation, errors involving technical or clerical problems, and so on. If the wrong sample is tested, for example, the subsequent events can be serious, such as a haemolytic transfusion reaction. Other errors may involve handing or storage, or be associated with different aspects of pre-transfusion testing. For example, the correct sample may be selected and tested but incorrect results recorded. Procedural errors may involve the incorrect test selection, resulting in wrong, inadequate, or inappropriate information being recorded. For example, recognition of mixed-field (double-cell populations) reactions can be important in preventing a wrong group being assigned. Manual techniques are more prone to errors than automated procedures, so it is recommended to install automated testing with good computer back-up where possible. Training remains imperative however, including understanding the equipment and software to be able to utilise the technology most effectively. Every laboratory must have an established internal incident reporting system as part of Total Quality Management. If team roles are not well-defined or understood, it could cause an error to occur due to lack of leadership, for example. Working conditions have been shown to contribute to error rates, as it has been shown that errors often occur out of normal working hours. Organisational factors include problems associated with staffing levels and other administrative details but in general, when an error occurs, it has many contributory elements and may not be due to a single factor. A process map indicating the step-by-step details can be helpful in analysing what should be changed, how and when a change should be implemented and by whom. This type of cause and effect analysis, followed by the appropriate corrective actions, will defend against possible error-prone acts or omissions, making each procedure as fool-proof as possible, aiming for zero errors and safe transfusion. However, sometimes discrepancies can occur, which must be resolved, often requiring further investigation at a reference laboratory. Rh typing anomalies may be resolved by reviewing the areas where a discrepancy may have occurred before referring the sample for further investigation as outlined in Table 9. Reference laboratories may perform additional tests in order to identify or confirm the presence of antibodies such as by the use of cord cells, lacking or having weak expression of certain antigens, or by the use of human body fluids known to neutralise certain antibodies. Autocontrol negative Varying strength of reactions, most screening and panel cells positive Anti-D due to antenatal or postnatal administration Comments Compatible blood easily found Compatible blood difficult to find. Depending on specificities, compatible blood may be difficult to obtain RhD negative cells to screen for other antibodies. This is useful in antibody mixtures of different immunoglobulin classes, to demonstrate potentially clinically important IgG antibody. The red cells from patients lack the Kx antigen due to deletion of part of the X chromosome. The McLeod phenotype exhibits greatly reduced expression of Kell antigens (see Chapter 5). Frequently asked questions C hapt er 1 0 What is the difference between sensitivity and specificity and how can these be determined Sensitivity is the number of positive results obtained, in comparison with the number of truly positive samples. Specificity is the number of negative results in comparison with truly negative samples. Anti-A,B was used to help in the detection of subgroups of A and B when only human, polyclonal antisera was available. It is now generally accepted that for routine blood grouping, monoclonal anti-A and -B reagents are sufficiently potent to detect such subgroups alone. Similarly, recommendations for reverse grouping have been reduced to A1 and B cells only. Ax, Am) may not be recognised using this format, but clinically, this is not considered important. The second test acts as a control against reagent malfunction or other technical errors.
