

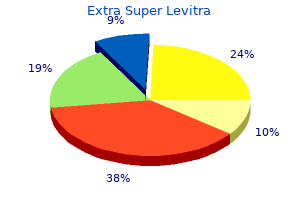
Purchase generic extra super levitra
A distinct tissue plane at the interface of tumour and adjacent parenchyma has been noted erectile dysfunction pills generic purchase cheapest extra super levitra and extra super levitra. The cell density is generally greater than that noted in normal grey matter, which is an important, yet inconstant, diagnostic feature. More importantly, these neuronal cells are spatially disordered, with no evidence of shared polarity, layering or respect for territory. Individual neurons possess large, vesicular nuclei, prominent and centrally positioned nucleoli, abundant cytoplasm with Nissl substance, and multipolar processes that are better visualized using silver stains or neurofilament immunohistochemistry. Cellular gigantism, coarse cytoplasmic vacuolisation and multinucleation are common. Gangliocytomas occasionally show evidence of neurofibrillary cytoskeletal changes, including well-defined tangles, as well as granulovacuolar degeneration and other neurodegenerative alterations. Some investigators suggest that nearly all ganglion cell tumours contain at least a minor neoplastic glial component and that true gangliocytomas are rare or nonexistent. The cytological anomalies present in the neuronal population are shared to some extent, although usually in a less pronounced form, by the abnormal neuronal elements in malformations of cortical development. Although gangliogliomas may be encountered at any age, 80 per cent present within the first three decades. They are most common in the cerebral hemispheres, with a strong predilection for the temporal lobes. Some observers report a relatively high incidence of bulbar and intramedullary examples in paediatric patients. Immunoreactivity for NeuN, an antigen present in nuclei of well-differentiated cortical neurons, is surprisingly negative to weakly positive in most ganglion cell tumours. The few ultrastructural investigations have emphasized neuronal dysmorphism and neurodegenerative features, depicting a variety of abnormal cytoplasmic inclusions. Cerebral examples are mostly associated with protracted, often medically refractory epilepsy. Gangliogliomas may account for over 20 per cent of all lesions involved in temporal lobe epilepsy resections, representing the most common neoplasm encountered in this clinical setting. Scalloping of the calvarium attests to the slow expansion of cerebral gangliogliomas, most of which are superficial (involving the cortical mantle). In other instances, these tumours may exhibit more limited cystic changes or may be entirely solid with tan or grey-white tissue. Most tumours are relatively demarcated, especially the cystic variants, and can acquire a firm texture owing to desmoplasia or a palpable grittiness due to calcification. Haemorrhage is rare and necrosis is mostly limited to previously treated or high-grade examples. Neuronal elements may be easily seen, occasionally dominating the histological picture, or are evident only after extensive searching, in some cases being sparsely distributed or regionally segregated. Whereas native ganglion cells are evenly distributed with orderly polarity and relatively unaltered cytology, neuronal cells in gangliogliomas usually lie in obvious architectural disarray, often clustering, and may exhibit pronounced dysmorphism. Chief among the latter are conspicuous variation in size and shape, multinucleation, cytoplasmic vacuolation, clumped Nissl substance and thickened, tortuous neuritic processes that sprout irregularly from cell bodies. Giant and bizarre forms that prove to be neuronal only on immunohistochemical assessment may be encountered, while some neurons bear neurofibrillary tangles and other abnormal cytoplasmic inclusions associated with neurodegenerative changes. Stromal fibrosis of gangliogliomas consists of a reticulin or collagenous network that can be minor and form wispy bridges between blood vessels or a more substantial spindle cell proliferation with fascicular or storiform patterns. Occasional solid gangliogliomas are less discrete and some fail to enhance or do so in a patchy or ring-like fashion. Some gangliogliomas feature extensive large vascular channels, resembling an arteriovenous malformation. Isolated mitoses, glial atypia (which may be pronounced, particularly in piloid regions), microvascular proliferation, leptomeningeal invasion (a common feature) and microscopic infiltration of adjoining brain tissue do 1730 Chapter 32 Neuronal and Mixed Neuronal-Glial Tumours not predictably affect outcome. These phenomena may prompt differential diagnostic consideration of desmoplastic infantile ganglioglioma or meningioma. In addition to highlighting neuronal cell bodies, antibodies to neurofilament proteins may delineate abnormal neuritic processes. Particularly striking in synaptophysin immunostains is the reaction pattern along perikaryal surfaces in a coarsely granular or linear fashion, a phenomenon that may reflect synapse formation and one that has been reproduced with antibodies to another synaptic vesicle-associated protein, synapsin I. Because such granules are not generally present in normal cortical neurons, they may be of diagnostic utility. Clear vesicles and synapses, including axosomatic contacts, may also be identified. Glial elements typically exhibit astrocytic features; their cell processes contain intermediate filaments and, in some cases, are covered by basal lamina where they abut the extracellular matrix. Oligodendrocyte-like cells with wellformed Golgi bodies, centrioles, mitochondria and microtubules may be identified. A rare instance of ependymal differentiation has been confirmed by ultrastructural demonstration of elaborate zonulae adherentes, microvilli and cilia. The histological picture may be indistinguishable, save for the presence of tumoural neurons, from that of glioblastoma. However, there are documented cases of anaplastic tumours faring well following complete excision, likely reflecting that some of these neoplasms retain compact, relatively non-infiltrative growth patterns amenable to surgical excision. In this regard, the reelin signalling pathway has been investigated for its potential involvement in the development of ganglioglioma. Recurrent partial imbalances comprised the minimal overlapping regions dim(10) (q25) and enh(12)(q13. Unsupervised cluster analysis of genomic profiles detected two major subgroups: 1) complete gain of 7 and additional gains of 5, 8 or 12; and 2) no major recurring imbalances or mainly losses. Their frequent association with developmental anomalies and typically indolent behaviour have prompted speculation that gangliogliomas represent tumoural forms of cortical dysplasia or benign neoplasms arising on a background of dysembryogenesis. Clonality studies have suggested a monoclonal origin for the glial and neuronal populations, but have not been conclusive. In fact, it is the large cystic component that is frequently responsible for the mass effect. Despite the large size, there is only mild oedema and communication with the ventricular system is rare. Their solid components lie largely outside the cerebrum proper, commonly being anchored to adjacent dura; they are grey-white and rubbery or firm owing to the large amounts of collagen. The tumour adheres to adjacent brain and these seemingly discrete neoplasms can demonstrate variable infiltration of neighbouring cerebral cortex. Epidemiology Desmoplastic infantile astrocytomas and gangliogliomas occur in the supratentorial compartment with the vast majority presenting in the first 2 years of life (mean age 6 months). Several reports have now described such tumours arising in older children, adolescents and young adults. The latter typically contain small cells of embryonal or astroglial appearance densely aggregated within a reticulin-free fibrillar matrix. Polygonal and gemistocytic cells may be seen in both fibrillar and desmoplastic regions. The presence of neuronal elements leads to the designation of desmoplastic infantile ganglioglioma rather than astrocytoma. Neuronal cells are most prevalent in the non-collagenous portions, range considerably in size and include ganglion cells that are fully differentiated, yet show dysmorphic features. Mitotic activity and necrosis are not conspicuous and are typically restricted to primitive small cell components. Examples with high mitotic rate, microvascular proliferation and necrosis have been documented, yet a more aggressive biology has not been demonstrated. There is also a modest population of fibroblasts with characteristically well-developed Golgi complexes and abundant rough endoplasmic reticulum that may be distended by granular material. Neuronal elements elaborate neuritic processes replete with microtubules, dense core granules and neurofilaments. In each case, either a normal karyotype or non-clonal abnormalities were described. The clinical, neuroimaging and histopathological features of central neurocytoma have been extensively reviewed. Cellular aggregates of undifferentiated and embryonal appearance are lacking in conventional ganglion cell tumours, although these may demonstrate considerable reticulin and collagen deposition.
Order extra super levitra 100mg line
Genital involvement significantly impairs sexual function and quality of life and may be overlooked if a specific examination and directed questions regarding genital symptoms are not undertaken safe erectile dysfunction pills generic 100 mg extra super levitra visa. Vulvo-vaginal involvement presents as erythema, erosions/fissures, vestibulitis, vaginal stenosis, labial resorption, or complete agglutination of the introitus leading to hematocolpos. The labia minora are partially resorbed with residual vulvitis and atrophic mucosa. Inflammation of the upper dermis is present, with extension of lymphocytes into the dermis and interface change. Sclerotic involvement of the upper dermis may resemble lichen sclerosus, with atrophy, hyperkeratosis, follicular plugging, and pale, homogenized appearance of the upper dermis collagen. Subcutaneous and fascial involvement accordingly demonstrates changes in the fat septae and fascia, including thickening, edema, and fibrosis. The inflammatory infiltrate is less dense than that usually seen in idiopathic lichen planus. There is hyalinization of the collagen throughout the dermis with loss of appendegeal structures. The presence of a normal leukocyte count is indicative of engraftment but no specific laboratory testing is diagnostic. Liver function testing and total bilirubin levels and quantification of diarrhea volume are used in conjunction with skin disease to stage the disease (Table 28-2). Sclerotic changes resulting in restriction in joint function lead to functional disability and joint contractures. Patients with mild (Grade I) skin involvement without hepatic or gastrointestinal symptoms may respond to high-potency topical steroids. However, more severe skin involvement or the presence of internal organ involvement necessitates treatment with systemic corticosteroids (methylprednisone 2 mg/kDa/day). Patients with skin sloughing require meticulous skin care, infection surveillance, and fluid management similar to toxic epidermal necrolysis. Approximately 50% of patients respond to systemic corticosteroids-however, those who require salvage therapy typically receive one or more immunosuppressive agents, including calcineurin inhibitors (tacrolimus, cyclosporine), mycophenolate mofetil, and sirolimus, which are of variable success (Box 28-5). Determination of a preferred second-line agent has been complicated by poor understanding of the disease process and a lack of high-quality clinical trials. Chapter 28 Ns100,140 Ns117 Ns142 Ns143,144 Ns22,145 Nsb89 Ns91 Ns98:: Graft-Versus-Host Disease Ns147,148 Ns149 Ns154 Sc: sclerotic skin disease; Ns: Nonsclerotic skin disease. Topical tacrolimus may not be tolerable at sites of significant inflammation or erosions. Phototherapy may be appropriate for patients with limited epidermal or sclerotic disease in whom systemic therapy is not otherwise warranted. Skin cancer risk assessment and concurrent use of photosensitizing medications should also be considered. Marcellus et al104 reported improvement in 20/27 evaluable patients with sclerotic disease treated with etretinate; however, six patients could not tolerate the treatment due to scaling or skin breakdown. Common side effects include peripheral and periorbital edema, myalgia, and fatigue. Generalized oral disease can significantly impair oral intake and quality of life and often result in the need for systemic intervention. Dental hygiene is very important in patients with decreased salivary function and home fluoride treatment is frequently recommended. If estrogen is not contraindicated, hormone replacement via topical cream, vaginal ring, or oral replacement may improve genital skin integrity. Limited vaginal scarring/synechiae can be treated with dilators or manual lysing; however, thick vaginal scarring may require surgical intervention. The benefits of T-cell depletion, however, are offset by higher rates of graft failure, cancer relapse, and infection. Couriel D et al: Ancillary therapy and supportive care of chronic graft-versus-host disease: National institutes of health consensus development project on criteria for clinical trials in chronic Graft-versus-host disease: V. Biol Blood Marrow Transplant 12(4):375-396, 2006 329 4 Chapter 29:: Skin Disease in Acute and Chronic Immunosuppression:: Benjamin D. In patients with acute immunosuppression, infections occur that are normally controlled by neutrophils and macrophages. In patients who have long-term immunosuppression, T-cell function is impaired and skin diseases are often similar to those seen in patients with human immunodeficiency virus infection. Salient dermatologic features particularly associated with immunosuppression are important diagnostic signs and indicators for therapy. While few skin conditions appear solely in immunocompromised individuals, clinical presentations may be morphologically atypical, follow unusual clinical courses, or prove harder to treat than in individuals with intact immunity. Other chapters cover graft-versus-host disease (see Chapter 28), skin signs associated with primary immunodeficiency disorders (see Chapter 143), and detailed side effects of medications, including corticosteroids, cancer chemotherapeutic agents, immunosuppressants, and cytokines (see Chapters 224, 227, 233, and 234). The salient clinical features particularly associated with immunosuppression are emphasized here. While a variety of inflammatory skin diseases and paraneoplastic processes occur in the setting of immunosuppression, infections, and malignancy are most commonly seen and are discussed herein. When approaching an immunocompromised patient, it is helpful to determine the time frame of the immune loss as well as the specific immune defect. This chapter is divided into two major subsections based on this concept: acute immunosuppression and chronic immunosuppression. When patients are acutely immunosuppressed, usually from iatrogenic ablation of the immune system or from acute leukemia, infections occur that are normally controlled by innate immunity, which typically involve neutrophils and macrophages. Thus, it is helpful to understand the underlying immune defects associated with the medical conditions of each patient (Table 29-1), because it helps to focus the history taking and physical examination toward skin manifestations of specific pathogens. Appropriate evaluation and diagnosis of skin lesions are critical to the overall health of these individuals, because the skin is often a window to more severe systemic illness. In particular, unusual presentations of infection with typical pathogens and infections with rare opportunistic pathogens are common in these patients. Diagnosis is also made more difficult by the variety of organisms that share similar morphologies and the wide variety of morphologic presentations of a single organism (Table 29-2). This makes prompt clinical evaluation and extensive use of skin biopsy and culture necessary to make an accurate diagnosis and initiate prompt treatment to obviate significant morbidity and mortality. Pancytopenia and neutropenia in particular predispose to invasive infections caused by gram-negative and -positive bacteria and the fungal organisms Candida and Aspergillus. In the past two decades, overall mortality due to infection among patients undergoing hematopoietic transplantation has decreased significantly with the use of better prophylaxis and nonmyeloablative regimens, but still represents an ongoing risk to survival. Muted clinical signs and symptoms can be found in this population, so care must be taken to rule out deeper involvement as occurs in necrotizing fasciitis. Bone marrow transplant patients and other patients with neutropenia are prone to streptococcal bacteremia and may develop facial flushing, a widespread erythematous, petechial or purpuric eruption of macules and papules, and desquamation of the palms and soles. Classically described in patients with Pseudomonas septicemia, it is now recognized that other bacterial and fungal organisms, including S. Patients with neutropenia, cystic fibrosis, or extensive burns are particularly susceptible to systemic P. Primary cutaneous infection, usually at the site of a medical procedure, can also cause ecthyma gangrenosum-like lesions. As is common with other infections in neutropenic patients, primary lesions can lead to bacteremia and should be treated aggressively. Mortality rates range from 40% to close to 100%, especially when treatment is delayed. Candidiasis and aspergillosis represent the two most common invasive fungal infections that occur in patients who are undergoing cytotoxic chemotherapy or stem cell transplantation or who have acute myeloproliferative disorders. Additional risk factors for opportunistic fungal infection include hyperalimentation, antibiotic use, hyperglycemia, corticosteroid use, and central venous catheter use. Other fungal organisms causing infection in hosts with acute neutropenia include Trichosporum species, Fusarium species, and organisms in the Zygomycetes class. Fungi may seed numerous organs, causing myositis, meningitis, endocarditis, pneumonitis, cerebritis, esophagitis, bursitis, osteomyelitis, arthritis, and endophthalmitis.
Extra super levitra 100mg free shipping
Ocular involvement in patients with cicatricial pemphigoid is common and may become sight threatening erectile dysfunction shakes menu order extra super levitra 100mg online. Although disease is usually bilateral, it often begins unilaterally and progresses to both eyes within several years. Patients may complain of burning, dryness, or a foreign-body sensation in one or both eyes; frank blisters on conjunctival surfaces are rarely seen. Because disease may be localized to the upper tarsal conjunctiva, it may escape detection without eversion of the eyelids. Chronic ocular involvement can result in scarring characterized by shortened fornices, symblepharons. Additional ocular complications include scarring of the lacrimal ducts, decreased tear secretion, and loss of mucosal goblet cells leading to decreased tear mucus content and unstable tear films. It is very important for patients with suspected ocular involvement to be examined by an ophthalmologist, because early disease may be subtle, is only identified by slit-lamp examination, and can result in severe complications. Other sites that may be affected by cicatricial pemphigoid include the nasopharyngeal, laryngeal, esophageal, and anogenital regions. Laryngeal involvement may present as hoarseness, sore throat, or loss of phonation. Chronic laryngeal erosions, edema, and scarring may result in supraglottic stenosis and airway compromise that eventually necessitates tracheostomy. Moreover, it has been suggested that esophageal dysfunction and gastroesophageal reflux may elicit or exacerbate laryngeal disease and/or bronchospasm in such patients. Although involvement of the genital and/or rectal mucosae in patients with cicatricial pemphigoid is rare, it can be a source of substantial pain and morbidity. Rare cases of urethral stricture, vaginal stenosis, and anal narrowing have developed as a consequence of this disease. Lesions typically consist of small vesicles or bullae situated on erythematous and/or urticarial bases. A cohort of 35 patients with antiepiligrin cicatricial pemphigoid (also called antilaminin 332 cicatricial pemphigoid) was shown to have an increased relative risk for cancer. The time between blister onset and cancer diagnosis was approximately 14 months in nine of the ten patients. This form of cicatricial pemphigoid appears to have a relative risk for malignancy that approximates that for adults with dermatomyositis; as is true for the latter, the risk for cancer appears to be particularly high in the first year of disease. Other patients with this form of cicatricial pemphigoid and cancer have been described more recently. In 1957, Brunsting and Perry described seven patients with locally recurrent and scarring subepidermal blistering lesions of the head or neck that for many years was thought to be a form of cicatricial pemphigoid. Light microscopy studies of older lesions often show fibroblast proliferation and lamellar fibrosis. Reports of patients with blisters in the sublamina densa region are thought to represent mucosapredominant forms of epidermolysis bullosa acquisita. In fact, this heterogeneity in autoantibody binding patterns was one of the first clues that cicatricial pemphigoid is a disease phenotype that is associated with different autoantigens (see Table 57-1). Although some studies have suggested that the use of human mucosal tissue substrates increases the likelihood of detecting autoantibodies in patients with cicatricial pemphigoid, other studies have not obtained similar results. Biopsy specimens as defined by requirements for medications to control disease as well as overall clinical severity score. Perilesional tissue from seronegative patients may be further characterized by immunoelectron microscopy studies to determine if in situ deposits of immunoreactants reside above or below the lamina densa of epidermal basement membrane. Distinguishing cicatricial pemphigoid from other autoimmune bullous diseases can be difficult and may require specialized immunopathologic studies and/or immunoelectron microscopy. Disorders that must be differentiated from cicatricial pemphigoid include lichen planus, erythema multiforme, lupus erythematosus, lichen sclerosus, and-in the case of ocular disease- cicatrizing or inflammatory conjunctivitis that results from long-term use of certain ophthalmologic preparations. These agents are particularly effective before bed, because oral secretions diminish during sleep. Because it is difficult to maintain contact of topical agents with mucous membranes (and because lesions often are localized to the gingiva), customized delivery trays to occlude topical glucocorticoids over lesional sites in the mouth are also useful. Mouthwash (dexamethasone 100 g/mL, 5 mL per rinse) used in a "swish-and-spit" regimen for 5 minutes two to three times each day represents another approach for topical therapy. For oral disease resistant to topical glucocorticoids, these agents can (in some instances) be administered intralesionally. In addition to these measures, patients should follow a strict regimen of oral hygiene that includes regular brushing, flossing, and cleaning of teeth. Cicatricial pemphigoid rarely goes into spontaneous remission; its treatment is largely determined by its severity and sites of involvement. Because of potentially severe complications, ocular, laryngeal, esophageal, and/or anogenital involvement requires aggressive management by teams of physicians familiar with specialized care of these organ systems. For severe disease affecting the ocular, pharyngeal, or urogenital epithelia, a combination of systemic glucocorticoids and an additional immunosuppressive is indicated. Such combined regimens have had success in halting the progression of severe ocular disease, limiting scarring, and producing long-term remissions. In an effort to avoid adverse effects and complications produced by prolonged treatment with immunosuppressive agents, some groups treat patients with intravenous immunoglobulin. Nasal lesions often benefit from twice-daily irrigation of the nasal passages with saline or tap water as well as the use of topical emollients. Esophageal involvement requires medical management to avert dysphagia, pain, tissue loss, and secondary complications such as gastroesophageal dysfunction and reflux, stricture formation, aspiration, laryngeal irritation, or bronchospastic pulmonary disease. All patients with cicatricial pemphigoid require long-term follow-up because of the possibility for this chronic disease to relapse. Kirtschig G et al: Interventions for mucous membrane pemphigoid/cicatricial pemphigoid and epidermolysis bullosa acquisita: A systematic literature review. Arch Dermatol 138:380, 2002:: Linear Immunoglobulin A Dermatosis and Chronic Bullous Chapter 58:: Linear Immunoglobulin A Dermatosis and Chronic Bullous Disease of Childhood:: Caroline L. Clinical presentations may mimic dermatitis herpetiformis, bullous pemphigoid, and cicatricial pemphigoid. May occur in association with inflammatory bowel diseases but only rarely associated with gluten sensitive enteropathy. Histology shows subepidermal collection of neutrophils at the basement membrane, often collecting in papillary tips with subepidermal blisters. Most patients respond dramatically to treatment with dapsone; some require adjunctive systemic corticosteroids. Linear immunoglobulin A (IgA) dermatosis is a rare immune-mediated blistering skin disease that is defined by the presence of homogeneous linear deposits of IgA at the cutaneous basement membrane. Clinical presentation of tense bullae, often in perineum and perioral regions, giving a "cluster of jewels" appearance. New lesions sometimes appear around the periphery of previous lesions with a collarette of blisters. Histology shows subepidermal collection of neutrophils at the basement membrane, similar to linear IgA bullous dermatosis. A minority of patients in both groups have additional deposits of other immunoreactants, most often IgG and occasionally the third component of complement (C3). Immunoelectron microscopy of the skin of patients with linear IgA deposits has revealed three distinct patterns of immunoreactants. Horiguchi et al reviewed 213 cases of linear IgA in Japan and found a strong association between older age of onset and both IgG/IgA type and dermal binding. Interestingly, when comparing the different groups based on patterns of antigen binding. Zone et al studied sera from patients who had circulating IgA antibodies that bound to the epidermal side of 1 M NaCl-split normal human skin, as shown by indirect immunofluorescence. Typically, these lesions are distributed symmetrically on extensor surfaces, including elbows, knees, and buttocks. Patients with drug-induced linear IgA bullous dermatosis have been reported with erythema multiformelike findings and a toxic epidermal necrolysis-like presentation, with widespread bullae.
Buy extra super levitra with a visa
Many drugs have been implicated in causing druginduced lupus syndromes erectile dysfunction doctors in ct purchase extra super levitra overnight delivery, especially hydralazine, procainamide, isoniazid, methyldopa, and minocycline. The number of patients who develop subacute cutaneous lupus erythematosus during treatment with these medications is very low, and these drugs are thought to have a low risk for causing or exacerbating cutaneous lupus. Cutaneous findings include livedo reticularis, painful nodules on the legs, and nondescript eruptions. A drug cause should be considered in the differential diagnosis of a wide spectrum of dermatologic diseases, particularly when the presentation or course is atypical. The diagnosis of a cutaneous drug eruption involves the precise characterization of reaction type. A wide variety of cutaneous drug-associated eruptions may also warn of associated internal toxicity (Table 41-3). Even the most minor cutaneous eruption should trigger a clinical review of systems, because the severity of systemic involvement does not necessarily mirror that of the skin manifestations. The most common serologic abnormality is positivity for antinuclear antibodies with a homogenous pattern. Fever, malaise, pharyngitis, and other systemic symptoms or signs should be investigated. A usual screen would include a full blood count, liver and renal function tests, and a urine analysis. Skin biopsy should be considered for all patients with potentially severe reactions, such as those with systemic symptoms, erythroderma, blistering, skin tenderness, purpura, or pustulation, as well as in cases in which the diagnosis is uncertain. Instead, diagnosis and assessment of cause involve analysis of a constellation of features such as timing of drug exposure and reaction onset, course of reaction with drug withdrawal or continuation, timing, and nature of a recurrent eruption on rechallenge, a history of a similar response to a crossreacting medication, and previous reports of similar reactions to the same medication. Several in vitro investigations can help to confirm causation in individual cases, but their exact sensitivity and specificity remain unclear. Investigations include the lymphocyte toxicity and lymphocyte transformation assays. Patch testing has greater sensitivity if performed over a previously involved area of skin. However, a reaction suggestive of a potentially life-threatening situation should prompt immediate discontinuation of the drug, along with discontinuation of any interacting drugs that may slow the elimination of the suspected causative agent. Antihistamines, topical corticosteroids, or both can be used to alleviate symptoms. Drug desensitization, also known as induction of drug tolerance, has been used primarily for IgE-mediated reactions caused by drugs such as penicillin or more recently, monoclonal antibodies such as rituximab and infliximab. However, once a reaction has occurred, it is important to prevent future similar reactions in the patient with the same drug or a cross-reacting medication. Some of these can be inherited, which places firstdegree relatives at a greater risk than the general population for a similar reaction to the same or a metabolically cross-reacting drug. Postmarketing voluntary reporting of rare, severe, or unusual reactions remains crucial to enhance the safe use of pharmaceutical agents. Eshki M et al: Twelve-year analysis of severe cases of drug reaction with eosinophilia and systemic symptoms. Mockenhaupt M: Severe drug-induced skin reactions: Clinical pattern, diagnostics and therapy. Justiniano H, Berlingeri-Ramos A, Sanchez J: Pattery analysis of drug-induced skin diseases. Most often begins as a single 2- to 4-cm thin oval plaque with a fine collarette of scale located inside the periphery of the plaque ("herald patch"). Similar-appearing, but smaller, lesions appear several days to weeks later, typically distributed along the lines of cleavage on the trunk ("Christmas tree" pattern). Treatment is usually supportive, although midpotency topical corticosteroids can reduce pruritus; high-dose acyclovir for 1 week may hasten recovery. The initial lesion is followed several days to weeks later by the appearance of numerous similar-appearing smaller lesions located along the lines of cleavage of the trunk (a so-called Christmas tree pattern). Patients are viremic, which may explain associated flu-like symptoms in some patients, and they generally do not have infected epithelial cells or large viral loads within skin lesions, which explains the difficulty in detecting these viruses by electron microscopy and by nonnested polymerase chain reaction testing. Children with roseola are viremic and skin lesions generally do not contain infected cells. As well, the characteristic distribution of lesions and differences in lesional and nonlesional skin are unexplained. In a minority of patients, flu-like symptoms have been reported, including general malaise, headache, nausea, loss of appetite, fever, and arthralgias. In approximately 20% of patients, the clinical picture diverges from the classic one described above. Localized forms of disease may involve colored, erythematous, or hyperpigmented (especially in individuals with darker skin); and demonstrates a fine collarette of scale just inside the periphery of the plaque. The primary plaque is usually located on the trunk in areas covered by clothes, but sometimes it is on the neck or proximal extremities. The interval between the appearance of the primary plaque and the secondary eruption can range from 2 days to 2 months, but the secondary eruption typically occurs within 2 weeks of the appearance of the primary plaque. The secondary eruption occurs in crops at intervals of a few days and reaches its maximum in approximately 10 days. The symmetric eruption is localized mainly to the trunk and adjacent regions of the neck and proximal extremities. The most pronounced lesions extend over the abdomen and anterior surface of the chest as well as over the back. Two main types of secondary lesions occur: (1) small plaques resembling the primary plaque in miniature, aligned with their long axes along lines of cleavage and distributed in a Christmas tree pattern, and (2) small, red, usually nonscaly papules that gradually increase in number and spread peripherally. The palms and soles are involved at times, and the clinical picture in these patients may simulate a widespread eczematous eruption. Typical histopathologic features include focal parakeratosis, a reduced or absent granular cell layer, mild acanthosis, mild spongiosis, papillary dermal edema, a perivascular and superficial dermal interstitial infiltrate of lymphocytes and histiocytes, and focal extravasation of erythrocytes. The histologic picture is indistinguishable from that of superficial gyrate erythema. In older lesions, the perivascular infiltrate is often both superficial and deep, with less spongiosis and more pronounced acanthosis. These late lesions may be difficult to distinguish from psoriasis and lichen planus. However, leukocytosis, neutrophilia, basophilia, lymphocytosis, and slight increases in erythrocyte sedimentation rate and levels of total protein, 1- and 2-globulins, and albumin have been reported. Pityriasis lichenoides chronica may present with a Christmas tree pattern on the trunk, but as a rule, typical lesions will be found on the extremities. Thus, it is always important to obtain a drug history to investigate this possibility. These include arsenic, barbiturates, bismuth, captopril, clonidine, gold, interferon-, isotretinoin, ketotifen, labetalol, organic mercurials, methoxypromazine, metronidazole, omeprazole, d-penicillamine, salvarsan, sulfasalazine, terbinafine, lithium, and tripelene amine hydrochloride. Nummular dermatitis: plaques usually circular and not oval, no collarettes of scale, tiny vesicles common. Pityriasis lichenoides chronica: longer disease course, smaller lesions, thicker scale, no herald patch, more common on extremities. The disease duration normally varies between 4 and 10 weeks, with the first few weeks associated with the most new inflammatory skin lesions and the greatest likelihood of flu-like symptoms. As with other skin diseases, this occurs more commonly in individuals with darker skin color, with hyperpigmentation predominating. Midpotency topical corticosteroids may be used for symptomatic relief of pruritus. For patients early in the disease course who demonstrate associated flu-like symptoms and/or extensive skin disease, oral acyclovir 800 mg five times daily for 1 week (or equivalent acyclovir derivative) may hasten recovery from disease. Watanabe T et al: Pityriasis rosea is associated with systemic active infection with both human herpesvirus-7 and human herpesvirus-6. Broccolo F et al: Additional evidence that pityriasis rosea is associated with reactivation of human herpesvirus-6 and -7. Drago F, Broccolo F, Rebora A: Pityriasis rosea: an update with a critical appraisal of its possible herpesviral etiology.
Order extra super levitra online
In children protein shakes erectile dysfunction discount extra super levitra 100mg otc, the brain stem and thalamus are characteristic locations for diffuse 1638 Overview and Biology of Diffuse Astrocytic Tumours 1639 astrocytomas. Pilocytic astrocytomas arise most frequently in the cerebellum, brain stem, optic pathways and hypothalamus. Non-localizing signs and symptoms are also common, particularly headaches, seizures and altered consciousness. In the setting of lower-grade lesions, seizures may be present for years before the onset of other clinical signs and symptoms. Ultimately, patients develop increased intracranial pressure owing to mass effect. Most glioma classifications have postulated that astrocytomas arise from astrocytes, and oligodendrogliomas from oligodendrocytes. Significant indicators of increasing grade in gliomas include cytological atypia, cellular density, mitotic activity, microvascular proliferation and necrosis. Importantly, both astrocytic differentiation and tumour grading are determined morphologically and subject to interpretation. A glioma progenitor cell could arise either from a stem cell population or from a differentiating progenitor cell population. Astrocytomas can develop through numerous pathways of acquired gene mutations, amplifications and deletions. For instance, overexpressing oncogenic Ras and Akt in progenitors results in mouse brain tumours that are histologically similar to glioblastomas, yet targeting more mature astrocytic progenitors is less oncogenic. Another gene related to chromatin remodeling, H3F3A, is mutated in astrocytomas and appears to be an early event, mostly in paediatric high-grade astrocytomas. There are two types of recurrent somatic mutations that occur in H3F3A, with one resulting in amino acid substitution at K27 and the other at G34. Mouse modelling studies have shown variations in tumour morphology depending on the type of cell transformed, as well as the oncogenic combination. Morphologically distinct regions in oligoastrocytomas have similar genetic alterations, indicating that these are clonal lesions, albeit with striking phenotypic diversity. Nonetheless, as more sensitive neuronal markers are utilized, many glial neoplasms seemingly coexpress neuronal and glial antigens, either uniformly or focally. It is interesting to note, however, that those oligodendroglial tumours with 1p and 19q loss preferentially affect particular areas of the brain, raising the possibility that specific precursor populations in different brain regions transform along distinct genetic pathways to reach common phenotypic end points, such as oligodendroglioma. Malignant glioma cells show preferential invasion along white matter tracts, around neurons and blood vessels, and in the subpial region. These infiltrative tendencies suggest that glioma cells have either a tropism for particular sites or a restricted ability to invade other regions. Moreover, glioma invasion is best viewed as the combined ability to migrate and to modulate the extracellular space. Unfortunately, investigations of glioma invasion have been hampered by a paucity of representative experimental models that mimic the human disease. In general, the extracellular matrix of the brain is ill defined and scant, consisting primarily of hyaluronic acid, except in two areas: around blood vessels and at the pial surface (glia limitans). At these sites, there is a well-defined and more traditional basal lamina that includes collagens. These degrade the extracellular environment to facilitate migration, but also remodel the environment in a manner that facilitates tumour cell growth. Studies of interactions between glioma cell surface molecules and extracellular matrix molecules have shown that gliomas express a variety of integrin receptors that mediate interactions with molecules in the extracellular space. The integrin heterodimers most clearly implicated have been 21 (interacting with tenascin), 51 (interacting with fibronectin), 61 (interacting with laminin) and v3 (interacting with vitronectin). Glioblastomas developing along this pathway have been termed secondary glioblastomas,215 in contrast to primary glioblastomas, which develop clinically de novo, i. Molecular changes underlying malignant progression of astrocytomas parallel histological changes and clinical course. At a molecular level, many genetic alterations target cell cycle regulatory genes. In the setting of rapidly dividing cells with high metabolic demands, regions of tumour distant from blood vessels may develop necrosis when metabolic demands exceed supply (diffusion-limited hypoxia and necrosis). Another consequence of hypoxia may be clonal selection of malignant cells that are able to survive selection pressure. Factors that initiate vaso-occlusion are not clear, yet endothelial cells of small vessels may be driven toward apoptosis and regression as a result of angiopoietin 2 expression. For example, hypoxia may act as a selective force that leads to the emergence of highly malignant and apoptosis-resistant tumour cells bearing inactivated p53. Studies in animal models indicate that these progenitors localize to angiogenic vessels in high-grade gliomas and are incorporated into their walls. Whether these cells are capable of transdifferentiating into endothelial cells or pericytes or have additional roles continues to be investigated. It manifests most frequently in young adults, with a peak incidence in the fourth and fifth decades. The diffuse increase in vascular density is not always an obvious feature on haematoxylin and eosin (H&E)-stained sections, but can be appreciated using immunohistochemistry for endothelial cells. Microvascular proliferation is a complex accumulation of proliferating endothelial and perivascular cells that lead to tufting and budding of the vasculature within neoplastic tissue. Most notably, complex glomeruloid bodies form semicircular garlands that hug regions of necrosis, highlighting the interrelationship between hypoxia/necrosis and angiogenesis. Less commonly, microvascular proliferation can be found at the invading edges, far from necrosis, and presumably as a result of angiogenic factors expressed by invading glioma cells. The biological underpinnings of microvascular proliferation are complex, with many angiogenic growth factors and their receptors found in glioblastomas. Infiltration often leads to enlargement and distortion, but not destruction, of involved grey or white matter. Normal nervous system cells and structures, including neurons, their axons, glial cells and blood vessels, are all typically entrapped within the lesion. Mitotic activity is generally absent, although isolated mitoses found after long searches of large resection specimens may not connote increased malignancy in the same way as finding a single mitosis in a needle biopsy. Neoplastic cell processes form a loose fibrillary matrix that is most often indiscernibly intertwined with the neuropil meshwork of the normal brain parenchyma. Stout, randomly oriented processes extend from cells and intermix with the normal neuropil. Transition to typical infiltrating astrocytoma is noted in the majority of cases, yet others consist almost entirely of atypical granular cells. Protoplasmic astrocytoma, a less common variant, has been described as containing neoplastic astrocytes with small cell bodies with few, thin processes and a low content of glial filaments. This lesion is not well defined and is considered by some to be a non-specific pattern, rather than a true variant. Note the gradient of increasing cellular density, which is helpful in establishing an infiltrative pattern of neoplasia. As with other infiltrating gliomas, there are often secondary structures, including subpial or subependymal condensation, perivascular aggregates, and perineuronal satellitosis. Several reports indicate that the gemistocytic variant appears particularly prone to malignant progression,121,176,256 whereas protoplasmic forms and tumours with extensive microcystic change may follow a slower course, often with a history of chronic seizures. The last group behaves very aggressively and is associated with much shorter survival times. However, the rim-like enhancement pattern typical of glioblastoma is not a feature. Nonetheless, there is infiltration without tissue destruction, often leading to enlargement of invaded structures. Rapid tumour growth and peritumoral oedema may lead to mass shifts and increased intracranial pressure. On cytologic preparations, the glial differentiation is usually more appreciable, with elongate cellular processes extending from individual neoplastic cells. Capillaries are lined by a single endothelial layer with frank microvascular proliferation, and necrosis is absent by definition.
Purchase genuine extra super levitra line
Typically erectile dysfunction labs discount extra super levitra 100mg free shipping, familial aggregation of cases exhibits a non-Mendelian pattern suggestive of polygenic, multifactorial inheritance. Its generally sporadic occurrence and unilateral distribution have led to the suggestion that it might result from somatic mosaicism for de novo mutations,14,15 perhaps in genes that are critical for melanoblast/melanocyte development or survival, although this hypothesis remains to be confirmed. Vitiligo can develop at any age,5 with a mean age-of-onset in Caucasian patients of about 24 years. Humoral immunity was first implicated by the finding in some cases of circulating antimelanocyte autoantibodies18 that target various melanocyte antigens, including tyrosinase, tyrosinase-related protein-1, dopachrome tautomerase, and others, and that have the capability to kill melanocytes in vitro19 and in vivo. As these T cells express a type-1 cytokine profile and colocalize with epidermal melanocytes, it has been hypothesized that these cells are actively cytolytic toward remaining melanocytes, via the granzyme/perforin pathway. Occupational vitiligo usually initially involves the hands and forearms (the site of contact with the inciting agent). At present, it is unclear whether these agents are directly toxic to melanocytes, or whether some individuals might be genetically susceptible to melanocyte injury from aliphatic phenolic derivatives, ultimately resulting in melanocyte death, release of antigenic intracellular proteins, loss of tolerance, and autoimmunity. On the basis of the polymorphic distribution, extension, and number of white patches, vitiligo is classified into generalized (vulgaris, acrofacial, mixed), universalis, and localized (focal, segmental, and mucosal) types. Mixed vitiligo-combination of acrofacial and vulgaris, or segmental and acrofacial types. Vitiligo universalis-complete or nearly complete depigmentation of the whole body. Focal vitiligo-characterized by the presence of one/few macule(s) in one area but not distributed in a segmental pattern. It generally affects young children and typically remains localized, the depigmented lesions persisting unchanged for many years. Various precipitating factors have been suggested, including physical trauma to the skin, sunburn, psychological stress, inflammation, pregnancy, contraceptives, vitamin deficiency, and many others. Quadrichrome vitiligo is characterized by the presence of a fourth color (dark brown) at sites of perifollicular repigmentation. Pentachrome vitiligo-the occurrence of five shades of color: (1) white, (2) tan, (3) medium brown, (4) dark brown, and (5) black. Vitiligolike depigmentation is thought to be a marker of the patient developing immunity against melanoma cells and to be an indicator of favorable prognosis, especially in advanced stages. Given the association between vitiligo and other autoimmune diseases, several screening laboratory tests are helpful, including T4 and thyroid-stimulating hormone levels, antinuclear antibodies, and complete blood count. Clinicians should also consider testing for serum antithyroglobulin and antithyroid peroxidase antibodies, particularly when patients have signs and symptoms suggestive of thyroid disease. Generally, histology shows an epidermis devoid of melanocytes in lesional areas,41 and sometimes sparse dermal, perivascular, and perifollicular lymphocytic infiltrates at the margins of early vitiligo lesions and active lesions, consistent with cell-mediated immune processes destroying melanocytes in situ. Further studies are needed to clarify this highly debated issue with obvious therapeutic implications. Ash leaf hypopigmented macules, facial/periungual angiofibromas, shagreen patches; autosomal dominant. Linear distribution, unilateral or bilateral pattern of hypopigmented macules; sporadic; chromosomal or genetic mosaicism. Hypopigmented lesions, beginning as reddish macules with fine scales upon scraping and seborrheic distribution. Depigmented round/oval patches (postinflammatory depigmentation) around the neck (necklace of Venus), trunk, limbs, or depigmented patches with peripheral reticular hyperpigmentation (primary lesion). Depigmented patches with polymorphic presentation, usually accompanied by localized anesthesia; histology: compact skin granulomas. Depigmentation at a distance from the tumor; the presence of tumor excludes typical vitiligo. Hypopigmenting inflammatory reactions leave ill-defined, poorly circumscribed lesions. Caused by use of systemic drugs (chloroquine, fluphenazine, physostigmine, imatinib, or topical imiquimod). Hypochromic pale lesion with well-defined borders and irregular margins; often solitary, located on the trunk. Histology and electron microscopic examination reveal no abnormality in melanocytes or melanization. Vitiligo repigmentation is assessed in terms of the proportion of treated subjects in whom a specified degree of repigmentation is achieved; depending on the study, more than 50% or more than 75% repigmentation may be considered a good response. Sometimes lesions spread over time, whereas in other cases disease activity stops, persisting in stable status for a long period. Spontaneous repigmentation is unpredictable, often clinically insignificant, and tends to be cosmetically unacceptable. Approximately 9 months of therapy are required to achieve maximal repigmentation; at least 3 months of treatment are warranted before the condition can be classified as nonresponsive. The most responsive sites are face, trunk, and limbs, and the least responsive sites are the hands and feet. Caution is necessary when using topical steroids on and around the eyelids, as their use can increase intraocular pressure and exacerbate glaucoma. Vitiligo recurrence after cessation of treatment and corticosteroid-induced side effects. Vitamin D derivatives are indicated for use in localized disease; benefits include lack of skin atrophy and their easy application. However, their role in vitiligo treatment remains controversial; whereas some studies have reported substantial benefit, others found vitamin D analogs ineffective. An advantage of this method is lack of scarring if recipient and donor sites are carefully manipulated. Next, minigrafts are harvested from the donor site using a similar punch and are transferred to recipient sites with fine forceps or a hypodermic needle. Good results are achieved in patients with refractory lip leukoderma, although the risk of cobblestoning seems to be high. These treatments produce optimal aesthetic results, with minor contrast between normal and affected skin. No data for cancer risk and other longterm side effects are available; therefore, caution is currently advised. An epidermal suspension collected from a small donor skin sample is prepared by 0. After 3 weeks, epidermal sheets are harvested from the culture vessel and transplanted onto depigmented recipient sites previously denuded by liquid nitrogen freezing, superficial dermabrasion, lasers, or diatermo surgery. A hyaluronic artificial matrix for growing keratinocytes and melanocytes has also been used with success. During subculturing, pigment cells increase in number and may be transplanted onto denuded areas at a density of up to 100,000 melanocytes/cm2. The suspension spreads onto recipient areas and is covered for a week, providing a good cellular take. Modern camouflage dyes and creams are waterproof, and the wide range of color and shades available can enable patients to choose the most suitable ones for their own skin color. Tanning should be avoided since it enhances the contrast of vitiligo lesions with normally pigmented skin. Moreover, sunscreens are needed to prevent sunburn of depigmented unprotected skin. However, this is somewhat problematic, as moderate sun exposure (heliotherapy) in many cases can induce epidermal repopulation with melanocytes. As well, it has been suggested that skin friction due to repeated application of sunscreen might exacerbate the disease,43 although there is no evidence to support this hypothesis. Consider psychiatric evaluation for patients with marked low self-esteem and depression.
Extra super levitra 100mg for sale
The processes of opsonization and phagocytosis are also mechanisms for another important function of complement-the clearance of immune complexes and apoptotic debris from the circulation erectile dysfunction diabetes pathophysiology buy extra super levitra 100 mg lowest price. Inflammation, characterized by vascular changes and ingress and activation of leukocytes and inflammatory proteins, serves to augment the localized immune response in tissue. Three mediators of inflammation initiated by complement activation are the complement fragments C3a, C4a, and C5a. These are called anaphylatoxins because of their ability to induce degranulation of mast cells. Receptors for C5a are expressed on endothelial cells, mast cells, eosinophils, basophils, monocytes, neutrophils, smooth muscle cells, and epithelial cells. Binding of C5a to endothelial cells results in increased vascular permeability and expression of P-selectin, both of which promote leukocyte accumulation in tissue. Binding to neutrophils results in increased neutrophil motility, adhesion to endothelial cells, and production of reactive oxygen species. The overall result is the accumulation of inflammatory cells at local sites in tissue where they can phagocytose and efficiently kill microbes. The activation of complement also results in stimulation of the humoral immune system through the generation of C3d. Opsonization by complement also facilitates antigen presentation to B cells by follicular dendritic cells. The alternative, classical, and lectin pathways converge at the formation of C5b and its sequential attachment to C6, C7, C8, and C9. The next steps do not involve enzymatic cleavage, but rather the sequential binding of C6, C7, and C8 to C5b. These pores may result in cell death through osmotic rupture, particularly in nonnucleated erythrocytes. Regulation of complement activation is provided by certain serum and cell surface proteins. Many of the regulatory cell surface proteins are expressed on human cells but not microbes, thus protecting human cells from complement damage. Some of the regulatory proteins downregulate complement activation by displacing components of the early steps of the cascade. It interacts with intercellular adhesion molecule 1 expressed endogenously on endothelial cells to stabilize the adhesion of leukocytes to endothelium, facilitating the recruitment of leukocytes from the circulation into tissue. The C5a receptor, which is a member of the seven-transmembrane -helical G-protein-coupled receptor family, was mentioned in the previous section. It can also serve as a downregulator of complement activation, as it is involved in the dissociation of C3 convertase complexes. Another major point of interaction of regulatory proteins is with bound C3b or C4b. For surface-bound C3b or C4b, inactivation occurs through the displacement of components of the alternative or classical pathway C3 convertase from C3b or C4b or through proteolysis of C3b or C4b. Factor H preferentially binds cell surfaces with high levels of sialic acid, and the relative abundance of sialic acid on human cells but not microbes further focuses the downregulation of complement activation on human cells. Proteolysis of C3b or C4b is mediated by factor I, a plasma protein that requires cofactors for its activity. Plasma S protein binds the C5b-7 complex and blocks its insertion into the cell membrane and also inhibits C9 polymerization. Genetic deficiencies of regulatory complement proteins may result in inappropriately prolonged complement activation, such as occurs with C1 inhibitor deficiency. Complement is associated with systemic lupus erythematosus through several possible mechanisms. These include increased risk of autoimmunity conferred by certain complement deficiencies, tissue damage resulting from autoantibody-induced complement activation, ineffective clearance of autoimmunity-promoting apoptotic debris, and failure to eliminate self-reactive B cells. Complement activation has been implicated in the pathogenesis of atherosclerosis, reperfusion injury after myocardial ischemia, diabetic microvascular disease, and cerebral infarct in ischemic stroke. Certain infectious agents have evolved mechanisms for evasion of destruction by complement, and some use complement receptors or regulatory proteins to gain entry into the cell. Patients with leukocyte adhesion deficiency-1 exhibit significant abnormalities of leukocyte adhesion and have recurrent infections (see Chapter 143). The risk for developing age-related macular degeneration is affected significantly by the presence of certain polymorphisms in genes of the complement system, particularly complement factor H but also factor B and C2. These observations underscore the protean roles of complement in the immune system. Complement activation has the potential for causing tissue injury, but complement components may be important in clearance of immune complexes and apoptotic debris. Apoptotic bodies that are not cleared effectively may be able to trigger autoimmunity through presentation of normally sequestered autoantigens to the immune system. The complement system has been subverted by certain infectious agents for entry into the cell. The deposition of C3b on the mycobacterial cell membrane leads to its uptake into macrophages, where it exists as an intracellular parasite. Knowledge of these evasive and subversive strategies of pathogens may be useful in designing vaccines and targeted therapies. Jung D et al: Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Acute urticaria/angioedema is caused by drugs, foods, occasionally infection in association with immunoglobulin E-dependent mechanisms (allergy), or metabolic factors. In the absence of urticaria, angioedema can be due to overproduction or impaired breakdown of bradykinin. Treatment of acute urticaria/angioedema relies on antihistamines and short courses of corticosteroids, and identification and elimination of endogenous and exogenous causes. Treatment of physical urticaria/angioedema includes high-dose antihistamine prophylaxis, except for delayed pressure urticaria. Treatment of chronic idiopathic or autoimmune urticaria/angioedema includes antihistamines (nonsedating preparations primarily), low-dose daily or alternate day corticosteroids, or cyclosporine. There is less pruritus (fewer type C nerve endings at the deeper cutaneous levels) but there may be pain or burning. Age, race, sex, occupation, geographic location, and season of the year may be implicated in urticaria and angioedema only insofar as they may contribute to exposure to an eliciting agent. In the National Ambulatory Medical Care Survey data from 1990 to 1997 in the United States, women accounted for 69% of patient visits. Most acute episodes are due to adverse reactions to medications or foods and in children, to viral illnesses. Episodes of urticaria/angioedema persisting beyond 6 weeks are considered chronic and are divided into two major subgroups: (1) chronic autoimmune urticaria (45%) and (2) chronic idiopathic urticaria (55%) with a combined incidence in the general population of 0. Various types of physical urticaria/angioedema may last for years, but the individual lesions last fewer than 2 hours (except delayed pressure urticaria) and are intermittent. Whereas 85% of children experience urticaria in the absence of angioedema, 40% of adult patients with urticaria also experience angioedema. Approximately 50% of patients with chronic urticaria (with or without angioedema) are free of lesions within 1 year, 65% within 3 years, and 85% within 5 years; fewer than 5% have lesions that last for more than 10 years. Angioedema alters the natural history, and only 25% of patients experience resolution of lesions within 1 year. The hereditary group is considered to be life long once the diagnosis becomes clinically manifest. They blanch with pressure as the dilated blood vessels are compressed, which also accounts for the central pallor of the wheal. The dilated blood vessels and increased permeability that characterize urticaria are present in the superficial dermis and involves the venular plexus in that location. Cutaneous mast cells, but not those from other sites, release histamine in response to compound 48/80, C5a, morphine, and codeine. Vascular permeability in skin is produced predominantly by H1 histamine receptors (85%); H2 histamine receptors account for the remaining 15%.
