

Cheap 50mg indomethacin otc
It crosses the placenta and is distributed into breast milk where concentrations higher than those in maternal plasma have been achieved rheumatoid arthritis test buy indomethacin 75mg lowest price. Atenolol undergoes little or no hepatic metabolism and more than 90% of that absorbed reaches the systemic circulation unaltered; it is excreted mainly in the urine. Sympathomimetics: severe hypertension with adrenaline and noradrenaline and possibly with dobutamine. Chlorphenamine, hydrocortisone and adrenaline should be immediately available in case of severe anaphylaxis. In most cases, immunisation develops within the first 15 days of treatment initiation. Patients presenting with immunisation show a faster decline in total but not active rabbit IgG levels. Aim to keep total lymphocyte count below 3% of total white cell count or 50 cells/L. Alternatively, keep absolute T cell count below 50 cells/L, and only dose when above this. The manufacturers advise that overdosage of Thymoglobuline may result in leucopenia (including lymphopenia and neutropenia) and/or thrombocytopenia. Avoid simultaneous transfusions of blood or blood derivatives and infusions of other solutions, particularly lipids. In this instance, concomitant use of heparin and hydrocortisone in an infusion solution of 0. Atorvastatin is metabolised by cytochrome P450 3A4 to ortho- and parahydroxylated derivatives and various beta-oxidation products. Atorvastatin is eliminated primarily in bile as active metabolites following hepatic and/ or extrahepatic metabolism, but does not appear to undergo significant enterohepatic recirculation. Antivirals: increased risk of myopathy with atazanavir, boceprevir (reduce atorvastatin dose), darunavir, fosamprenavir, indinavir, lopinavir, ritonavir, saquinavir or tipranavir (max dose of atorvastatin 10 mg); concentration reduced by efavirenz and possibly etravirine; avoid with telaprevir. Lipid lowering agents: increased risk of myopathy with fibrates, gemfibrozil (avoid) and nicotinic acid. Rhabdomyolysis with renal dysfunction secondary to myoglobinaemia has been reported with other statins. The presence of food, particularly high fat food, increases bioavailability 2 or 3-fold. The most commonly reported abnormalities in laboratory parameters are increased liver function tests and amylase levels, and hyponatraemia. Antibacterials: aminoglycosides, clindamycin, polymyxin, piperacillin enhance effect of atracurium. Atracurium enhances the neuromuscular block produced by botulinum toxin (risk of toxicity). Antineoplastic agent: Treatment of people not eligible for stem cell transplants with myelodysplastic syndromes, chronic myelomonocytic leukaemia or acute myeloid leukaemia. If unexplained reductions in serum bicarbonate levels <20 mmol/L occur, the dose should be reduced by 50% on the next cycle. Patients with renal impairment should be closely monitored for toxicity since azacitidine and/or its metabolites are primarily excreted by the kidney. Mercaptopurine is rapidly and extensively metabolised in the liver, by methylation, oxidation and by the formation of inorganic sulfates. About 10% of a dose of azathioprine is reported to be split between the sulfur and the purine ring to give 1-methyl-4-nitro-5-thioimidazole. Metabolites and small amounts of unchanged azathioprine and mercaptopurine are eliminated in the urine. Cytotoxic agents may be additive or synergistic in producing toxicity, particularly on the bone marrow. Approximately 55% of radioactivity was recovered in faeces and approximately 42% in urine, with 15% of the dose excreted in urine as azilsartan. Ciclosporin: may inhibit the metabolism of ciclosporin (increased ciclosporin levels). May be used safely in patients on tacrolimus who require treatment with a macrolide. Aztreonam is excreted as unchanged drug with only small quantities of metabolites, mainly in the urine, by renal tubular secretion and glomerular filtration. Deamination yields the main metabolite, -(p-chlorophenyl)4-hydroxybutyric acid, which is pharmacologically inactive. Baclofen can be given intrathecally (at doses greatly reduced compared with oral dose), by bolus injection, or continuous infusion. Use with caution as a case report of encephalopathy has been reported in a haemodialysis patient. It is broken down by the colonic bacterial flora into 5-aminosalicylic acid (mesalazine), which is active, and 4-aminobenzoylalanine, which is considered to be an inert carrier. Most of a dose is eliminated via the faeces, but about 25% of the released mesalazine is absorbed and acetylated. A small proportion of 4-aminobenzoylalanine is absorbed and acetylated by first-pass metabolism through the liver. Mesalazine is best avoided in patients with established renal impairment, but if necessary should be used with caution and the patient carefully monitored. No data Potentially hazardous interactions with other drugs Ciclosporin: may alter ciclosporin requirements. It is most likely removed by opsonisation via the reticuloendothelial system when bound to lymphocytes, or by human antimurine antibody production. Use with caution in patients who have previously had basiliximab due to increased risk of developing hypersensitivity reactions. There was a trend toward higher clearance of belatacept with increasing body weight. Although proteinuria (2 g/day) increased belimumab clearance and decreases in creatinine clearance decreased belimumab clearance, these effects were within the expected range of variability. Another major route of bendamustine metabolism involves conjugation with glutathione. About 30% is excreted unchanged in the urine with the remainder excreted as uncharacterised metabolites. Anti-arrhythmics: hypokalaemia leads to increased cardiac toxicity; effects of lidocaine and mexiletine antagonised.
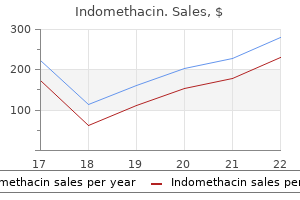
Best buy for indomethacin
A few days later the test result comes back and reveals that Luke has a deletion of exons 44 to 47 arthritis toes buy cheap indomethacin 25 mg, which has resulted in juxtaposition of out-of-frame exons. Case 6: Targeted treatment Mary is a 63 year old referred for evaluation of fatigue and weight loss. She had been healthy until about 3 months ago, but now has little energy and has lost 15 pounds (7 kg). On examination she is noted to have a palpable spleen and slightly enlarged liver. Blood testing is done and she is found to have 22 000 white blood cells per cubic millimetre, and a mild anaemia. This explains the enlarged liver and spleen, which are probably similarly engorged with myeloid cells. A Self-assessment case studies: questions 201 bone marrow aspirate is done, and chromosomal analysis reveals the presence of the Philadelphia chromosome. After consideration of her options, it is decided to start her on treatment with imatinib. Mary has been on treatment for 2 months, and her white blood cell count has returned to normal. Mary has been doing well for 18 months, but recently she has begun to feel fatigued again. She is found once again to have increased white blood cells on her blood smear, and Philadelphia chromosome positive cells are once again found. Case 8: A sleepy infant You are called to see Kirsten, a 1-day-old girl, in the newborn intensive care unit. Her doctors are concerned because she seems to be unusually sleepy, with minimal responses to stimulation and very lethargic feeding. Evaluation for infection has been negative, and she has no evidence of any metabolic derangement. She is very lethargic and hypotonic, though there are no fasciculations (visible muscle flickering) and deep tendon reflexes can be elicited, which both argue against spinal muscular atrophy. The extreme lethargy and poor feeding are more typical of a central nervous system, rather than a neuromuscular cause for the hypotonia, though congenital myopathy or congenital myotonic dystrophy remain a possibility. They ask whether they would be at risk of having another child with the disorder, and whether their healthy 6 year old will be at risk of having affected children when she grows up. Case 7: Worries about senility Larry is a 54 year old whom you have been treating for mild hypertension for the past 4 years. His health is otherwise good, though he also has hypercholesterolaemia, for which he was recently started on statin therapy to protect him from atherosclerosis by lowering his cholesterol. His mother had been diagnosed as having presenile dementia several years ago, and recently died of the disorder. Larry is told that there is no genetic testing that would be recommended at this time. Not entirely satisfied, Larry goes on the internet that night and does some research on his own for genetic testing for Alzheimer disease. You explain that this testing is not recommended as a screen for Alzheimer disease. His wife helps him do the cheek brushing, and the sample is sent off to the laboratory. A month later, Larry gets a report in the mail saying that his ApoE genotype is 2/4. They are not aware of a family history of any specific genetic disorder and both are in good health. Case 11: Autism spectrum disorder Jake is 4 years old and his parents have been increasingly concerned about him. Although he has achieved more or less normal motor developmental milestones, his speech and social development have been very slow. He spends most of his time playing on his own, with very little interaction with others, either his parents, his sibs, or other children. Case 10: Enzyme replacement Tom is a 41 year old who is seen in the Nephrology Clinic. At that time, he had suddenly developed left-sided weakness and diplopia (double vision), and was diagnosed as having had a mild stroke. Over the ensuing years he has noted that he does not sweat and has had some difficulty with overheating in hot weather. A family history is taken; no one on either side of the family had ever been diagnosed with autism. Fragile X results were normal, but Jake was found to have a deletion involving chromosome 16, at 16p11. His brother is also known to be affected and is undergoing enzyme replacement therapy. Tom is wondering whether he might also be a candidate for enzyme replacement therapy. His physical examination is notable for angiokeratoma on the palms of the hands and both knees. He has mild weakness in his legs and some decreased sensation to light touch in the fingertips and toes. Ophthalmological examination reveals bilateral corneal opacities and dilated conjunctival vessels, although his vision is normal. It is determined that Tom is eligible for enzyme replacement therapy, and when his initial evaluation is completed he is introduced to the protocol. His energy level has increased and he reports marked decrease in the pain in his fingers and toes. Case 12: Exome sequencing Zoe is 12 years old and has a long history of intellectual disability and congenital anomalies. She had a congenital heart defect that was repaired when she was an infant, as well as a cleft lip that was also surgically repaired. She was of normal birth weight, but has grown slowly since infancy and is now less than 5 feet tall. Over the years she has been seen by many specialists, but in spite of many genetic tests, no diagnosis has been achieved. Recently her paediatrician referred her back to a geneticist, who is unable to make a specific clinical diagnosis, but suggests that whole exome sequencing be done. A letter is sent to their insurance company, which agrees to cover the costs of testing. They are particularly glad to know that Zoe is affected by new mutation, since neither of them was found to carry the same mutation. She has done well for the most part, but has required hospitalized for pulmonary infections on several occasions. During one of her routine visits her physician speaks with her parents about a recent clinical trial that resulted in development of a new medication that is now approved for treatment of cystic fibrosis. Marci is tested and indeed has one copy of the Gly551Asp mutation (her other mutation is Phe508del, i. She is monitored for side effects, and after 6 months of treatment has tolerated the medication well. She also has experienced an improvement in pulmonary function, though she still requires treatment with antibiotics and chest physical therapy. He had heard at a party about the possibility of arranging genomic testing on himself and after reading the information on the website, decided to go ahead.
Syndromes
- Lightheadedness when standing
- Low level of sexual interest and impotence
- Bone marrow aspiration and biopsy
- Mental status changes
- When did the bed wetting begin? How often does the bed wetting occur? Have there ever been "dry" periods?
- Reactions to medicines
- The type of spider
- Cold cuts, hot dogs, salami, or sausage
- Turn into dark red, tender nodules
- Tube through the mouth into the stomach to empty the stomach
Generic 25 mg indomethacin with amex
Right arthritis in fingers and knees order indomethacin 25mg line, Note the relatively severe thumb (radial) defect of the right hand and the much more subtle "radial" defect of the left hand (arrow). The fallopian tubes and ovaries are usually nearly normal with normal secondary sexual characteristics, except for a lack of menstruation. The lower vagina, which is derived from an outpouching from the urogenital sinus, is usually present as a blindly ending pouch. Although most cases are sporadic, approximately 4% of cases have been familial, with affected female siblings. Absence of proximal two thirds of vagina and absence to hypoplasia of uterus (96%, but there is an ascertainment bias for this defect; sometimes referred to as the Rokitansky malformation sequence); renal agenesis or ectopy (88%). Infrequent features include deafness, cerebellar cyst, external ear defects, facial asymmetry, cleft lip and palate, micrognathia, gastrointestinal defects, anorectal malformations, defects of laterality, abnormal lung lobation, and occipital encephalocele. When interpreting a structural defect, the clinician is looking back to an early stage in development with which he or she has often had little acquaintance. This chapter sets forth some of the phenomena of morphogenesis and the normal stages in early human development, followed by the types of abnormal morphogenesis and the relative timing of particular malformations. Interaction between Adjacent Tissues the optic cup induces the morphogenesis of the lens from the overlying ectoderm, the ureteric bud gives rise to the development of the kidney from the adjacent metanephric tissue, the notochord is essential for normal development of the overlying neural tissue, and the prechordal mesoderm is important for the normal morphogenesis of the overlying forebrain. These are but a few examples of the many interactions that are essential features in morphogenesis. After the first few cell divisions, differentiation begins to take place, presumably through activation or inactivation of particular genes, allowing cells to assume diverse roles. The entire process is programmed in a timely and sequential order with little allowance for error, especially in early morphogenesis. Although little is known about the fundamental processes that control morphogenesis, it is worthwhile to mention some of the normal phenomena that occur and to give examples of each. Adhesive Association of Like Cells In the development of a structure such as long bone, the early cells tend to aggregate closely in condensations, a membrane comes to surround them, and only later do they resemble cartilage cells. The association of like cells is dramatically demonstrated by admixing trypsinized liver and kidney cells in vitro and observing them reaggregate with their own kind. Examples include death of tissue between the digits resulting in separation of the fingers and recanalization of the duodenum. The dead cellular debris is engulfed by large macrophages, leaving no trace of the tissue. Cell Migration the proper migration of cells to a predestined location is critical in the development of many structures. For example, the germ cells move from the yolk sac endoderm to the urogenital ridge, where they interact with other cells to form the gonad. Hormonal Influence over Morphogenesis Androgen effect is one example of a hormonal influence over morphogenesis-in this case, that of the external genitalia. Control over Mitotic Rate the size of particular structures, as well as their form, is largely the consequence of control over the rates of cell division. The size, growth, and form of the brain and its early derivatives, for example, have a major function in the formation of the calvarium and upper face. The alignment of collagen fibrils and bone trabeculae relates directly to the direction of forces exerted on these tissues. The first week is a period of cell division without much enlargement, the conceptus being dependent on the cytoplasm of the ova for most of its metabolic needs. By 7 to 8 days, the zona pellucida is gone, and the outlying trophoblast cells invade the endometrium and form the early placenta that must function both to nourish the parasitic embryo and to maintain the pregnancy via its endocrine function. During this time, a relatively small inner cell mass has become a bilaminar disk of ectoderm and endoderm, each with its own fluid-filled cavity, the amniotic sac and yolk sac, respectively. By the end of the second week, a small mound, a primitive node, has developed in the ectoderm, and behind it a primitive streak forms. Ectodermal cells migrate through the node and the primitive streak to specific areas between the ectoderm and endoderm, becoming the mesoderm. One of the early mesodermal derivatives is a circulatory system: During the third week, the heart begins to develop, vascular channels form in situ, and blood cells are produced in the yolk sac. The major part of the conceptus, the cytotrophoblast, has invaded the endometrium, and the embryo (arrow) is differentiating into two diverse cell layers, the ectoderm and endoderm. The thicker ectoderm (arrow) has its continuous amniotic sac, whereas the underlying endoderm has its yolk sac. Mesoblast cells migrate from the ectoderm through the node (the hillock marked by the arrow) and the primitive streak to specific locations between the ectoderm and endoderm, constituting the highly versatile mesoderm. Anterior to the node the notochordal process develops, providing axial support and influencing subsequent development such as that of the overlying neural plate. The stage is now set for the period of major organogenesis, which is best considered in relation to individual structures. As noted in the illustrations found on the inside front cover and inside back cover of this book, each stage of development represents a synchronous syndrome of characteristics. The second is deformation, caused by altered mechanical forces on a normal tissue. Deformation may be secondary to extrinsic forces, such as uterine constraint on a normal fetus, or to intrinsic forces related to a more primary malformation. The third type of pathology is disruption, which is a result of the breakdown of previously normal tissue. The midaxial ectoderm has thickened and formed the neural groove (arrow), partially influenced by the underlying notochordal plate (N). Vascular channels are developing in situ from mesoderm, blood cells are being produced in the yolk sac wall, and the early heart is beating. The fore part of the embryo is growing rapidly, especially the anterior neural plate. The cardiac tube (long arrow), under the developing face (short arrow), is functional. For example, most minor anomalies represent deformations, often secondary to a malformation. Malformations may be broken down into a number of subcategories in terms of the nature of the poor formation. Types of Malformation Incomplete Morphogenesis these are anomalies that represent incomplete stages in the development of a structure; they include the following subcategories, with one example listed for each. This dorsal view beautifully shows the developing brain (anterior) and spinal cord just after neural tube formation and the orderly bilateral segmentation of the somites. Such an anomaly may be more specific for a particular clinical syndrome entity than anomalies of incomplete morphogenesis. The fourth mechanism of abnormal morphogenesis is dysplasia, in which there is a lack of normal organization of cells into tissue. These anomalies represent an organizational defect leading to an abnormal admixture of tissues, often with a tumor-like excess of one or more tissues. Examples of hamartomas are hemangiomas, melanomas, fibromas, lipomas, adenomas, and some strange admixtures that defy traditional classification. A few disruption patterns of anomaly are considered in this book, as well as some dysplasias, with the major emphasis being on patterns of malformation, including malformation sequences. However, Accessory Tissue Accessory tissue such as polydactyly, preauricular skin tags, and accessory spleens may be presumed to have been initiated at approximately the same time as the normal tissue, developing into finger rays, auricular hillocks of His, and spleen, respectively. Functional Defects Function is a necessary feature in joint development; hence, joint contractures, such as clubfoot, may be caused by a functional deficit in the use of the lower limb resulting from a more primary malformation. Scanning electron microscope photograph of a 28- to 30-day-old human embryo with the amnion removed, showing the following features: 1, early optic vesicle outpouching; 2, maxillary swelling; 3, mandibular swelling; 4, hyoid swelling; 5, heart; 6, somites, with adjacent spinal cord; 7, early rudiments of upper limb bud; and 8, tail. Between it and the mandibular process is the area of the future mouth, where the buccopharyngeal membrane, with no intervening mesoderm, has broken down. Within the recess of the mandibular (M) and hyoid (H) processes, the future external auditory meatus will develop (arrow), and dorsal to it the otic vesicle (O) forms the inner ear. The relatively huge heart must pump blood in the yolk sac and developing placenta as well as to the embryo proper. Foregut outpouchings and evaginations will now begin to form various glands and the lung and liver primordia. The somites, which will differentiate into myotomes (musculature), dermatomes (subcutaneous tissue), and sclerotomes (vertebrae), are evident on into the tail bud.
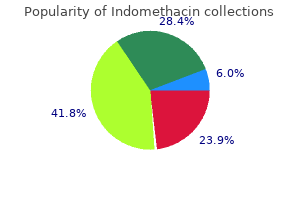
Purchase cheap indomethacin online
This is a very rare condition; due to failed production of red blood cell precursors in the bone marrow arthritis definition best order indomethacin, resulting in profound anaemia. Resulting antibodies render the patient unresponsive to the therapeutic effects of all epoetins and darbepoetin. A second metabolite, 6,15-diketo-13,14-dihydroprostaglandin F1, is formed by enzymatic degradation. Following the administration of radiolabelled epoprostenol to humans, at least 16 metabolites were found, 10 of which were structurally identified. Unlike many other prostaglandins, epoprostenol is not metabolised during passage through the pulmonary circulation. Some patients may exhibit allergic reaction to buffer solution used to reconstitute epoprostenol. The concentrated solution should be filtered using the filter provided in the pack. In the urine, approximately 20% of the radioactivity excreted was an acyl glucuronide of eprosartan with the remaining 80% being unchanged eprosartan. Following intravenous [14C] eprosartan, about 61% of radioactivity is recovered in the faeces and about 37% in the urine. Following an oral dose of [14C] eprosartan, about 90% of radioactivity is recovered in the faeces and about 7% in the urine. Reduce infusion to 1 mcg/kg/minute and use with caution due to limited experience. However, it is unknown whether Pgp is contributing to the biliary excretion of eribulin. A study in patients with different degrees of impaired renal function showed that the exposure of eribulin in patients with creatinine clearance 40 to 59 mL/min, n=6) was similar to patients with normal renal function while the exposure in patients with creatinine clearance <40 mL/ min was increased by 75%, n=4. They are present in plasma at levels that are <10% of erlotinib and display similar pharmacokinetics as erlotinib. Erlotinib is excreted predominantly as metabolites via the faeces (>90%) with renal elimination accounting for only a small amount (approximately 9%) of an oral dose. Antacids: concentration possibly reduced by antacids, give at least 4 hours before or 2 hours after erlotinib. Dilute solutions are stable for 6 hours at room temperature or 24 hours in a refrigerator. The major metabolite of ertapenem is the ring-opened derivative formed by dehydropeptidase-I-mediated hydrolysis of the beta-lactam ring. Of the 80% recovered in urine, approximately 38% is excreted as unchanged ertapenem and approximately 37% as the ring-opened metabolite. Anecdotally ertapenem has been used at a dose of 1 g 3 times a week in haemodialysis patients. Give at least 6 hours before haemodialysis session if unable to give post dialysis. It is excreted in high concentrations in the bile and undergoes intestinal reabsorption. Anxiolytics and hypnotics: inhibits midazolam and zopiclone metabolism; increases buspirone concentration. Atomoxetine: increased risk of ventricular arrhythmias with parenteral erythromycin. Calcium-channel blockers: possibly inhibit metabolism of calcium-channel blockers; avoid concomitant use with lercanidipine. Monitor blood levels of ciclosporin carefully and adjust dose promptly as necessary. Lipid-lowering drugs: increased risk of myopathy; concentration of rosuvastatin reduced; avoid concomitant use with simvastatin. Monitor blood levels of tacrolimus carefully and adjust dose promptly as necessary. Theophylline: inhibits theophylline metabolism; if erythromycin given orally decreased erythromycin concentration. Escitalopram and major metabolites are assumed to be eliminated by both hepatic and renal routes, with the major part of the dose excreted as metabolites in the urine. Minor metabolites in plasma are R-licarbazepine and oxcarbazepine, which were shown to be active, and the glucuronic acid conjugates of eslicarbazepine acetate, eslicarbazepine, R-licarbazepine and oxcarbazepine. Eslicarbazepine acetate and its metabolites are mainly excreted in the urine unchanged. This occurs through hydrolysis of the ester group by esterases in the red blood cells. Esmolol hydrochloride is excreted by the kidneys, partly unchanged (less than 2% of the administered amount), partly as acid metabolite that has a weak (less than 0. Antihypertensives: enhanced hypotensive effect; increased risk of withdrawal hypertension with clonidine; increased risk of first dose hypotensive effect with post-synaptic alpha-blockers. Sympathomimetics: severe hypertension with adrenaline and noradrenaline and possibly dobutamine. Almost 80% of an oral dose of esomeprazole is excreted as metabolites in the urine, the remainder in the faeces. Antifungals: absorption of itraconazole and ketoconazole reduced; avoid with posaconazole; concentration possibly increased by voriconazole. Stir well until it disintegrates; the liquid with pellets should be drunk immediately or within 30 minutes of preparation. Some hydrolysis of the carbamate linkage occurs in the liver, releasing estradiol, estrone, and the normustine group. Estramustine and estromustine are excreted with their metabolites mainly in the faeces. Pharmacokinetics of estramustine phosphate (Estracyt) in prostatic cancer patients. Dosages should be individually determined and adjusted according to measured levels and renal replacement therapy. Daily dosing is preferred by some specialists to aid compliance and ensure maximum therapeutic effect. Dose in renal impairment is from Drug Prescribing in Renal Failure, 5th edition, by Aronoff et al. Anti-epileptics: concentration possibly reduced by carbamazepine, phenytoin and phenobarbital; concentration of phenytoin possibly increased; concentration increased by valproate. The dose should be kept as low as possible and renal function should be monitored. Pharmacokinetics are complex and have been described by both 2- and 3-compartment models.
Purchase cheap indomethacin line
Normal cranial ossification; short ribs without fractures; short rheumatoid arthritis definition and causes discount indomethacin 75mg, broad long bones with disproportionately long fibula and metaphyseal irregularity of distal ulna; variable degrees of failure of ossification of lumbar spine, cervical spine, sacrum, ischial and pubic bones, and calcaneus and talus. In all cases where mutations have been identified, they have been heterozygous, indicating an autosomal dominant mode of inheritance. A number of examples of more than one affected child in a family born to unaffected parents have been reported and are felt to be the result of germline mosaicism. Wainwright H, Beighton P: Visceral manifestations of hypochondrogenesis, Virchows Arch 453:203, 2008. Note the relatively normal cranial ossification, short ribs, and variable degrees of failure of ossification of lumbar and cervical spines, sacrum, and ischial and pubic bones. A distinctive fibrosis of the growth-plate cartilage led to the designation fibrochondrogenesis. Widely patent anterior fontanel, coronal and sagittal sutures; protuberant eyes with large corneae; hypoplastic nose with flat nasal bridge and anteverted nares; long philtrum; small mouth; cleft palate; short neck; low-set, malformed ears. Flattened vertebrae with posterior vertebral hypoplasia and a sagittal midline cleft; short, thin ribs with anterior and posterior cupping; long, thin clavicles; small chest; small/elevated scapula. Rhizomelic shortening; small hands and feet; camptodactyly; fifth-finger clinodactyly; hypoplastic finger and toenails; short, dumbbellshaped long bones with broad, irregular metaphyses; prominent metaphyseal spurs adjacent to growth plates; short fibulae. Hypoplastic with ovoid ilia, irregular flattened acetabula with medial spikes and narrow sacrosciatic notches; broad, hypoplastic ischii. References Lazzaroni-Fossati F, et al: La fibrochondrogenese, Arch Fr Pediatr 35:1096, 1978. Bankier A, et al: Fibrochondrogenesis in male twins at 24 weeks gestation, Am J Med Genet 38:95, 1991. Note the flat, wide nasal bridge, anteverted nares, short limbs, and equinovarus position of the feet. B, the radiograph reveals long, thin clavicles; short, thin ribs; flattened acetabula; narrow sacrosciatic notches; metaphyseal widening of the tibia and fibula; and dumbbell-shaped femora. There is shortening of proximal and distal bones of the limbs, radial deviation of the thumb, and a large gap between toes 1 and 2. Neonatal death is uniform and related to pulmonary hypoplasia, tracheobronchomalacia, and a malformed stenotic larynx. Radiographic features include platyspondyly, cervical kyphosis, hypoplasia/dysplasia of vertebrae, short ribs, glenoid hypoplasia, horizontal acetabulae, bifid or V-shaped humerus, rounded distal femora, bowing of radius and tibia, and hypoplasia/dysplasia of tubular bones of hands and feet. Inactivation of the gene product, a sulfate-chloride exchanger of the cell membrane, leads to intracellular sulfate depletion and to synthesis of undersulfated proteoglycans in susceptible cells. This early lethal short-limbed dwarfing condition was set forth by Maroteaux and colleagues and Sillence and colleagues. Atelosteogenesis derives from the Greek word for "incomplete" and relates to the marked lack of complete ossification of certain bones. Humeri are absent, segment-shaped, or distally tapered; absent fibula; short distally pointed femora; bowed tibiae; abnormally segmented and fused cervical vertebrae; thoracic platyspondyly with multiple coronal clefts throughout; 11 pairs of ribs; narrow thoracic cage; hypoplasia of ischiopubis and flared ilia; lack of ossification of calcaneal centers; markedly delayed ossification of proximal phalanges and middle phalanges with well-ossified distal phalanges. Ocular hypertelorism, depressed nasal bridge, midface hypoplasia, micrognathia, multiple large joint dislocations, talipes equinovarus, polyhydramnios. Low-set ears, helix hypoplasia, and stenotic ear canals, hypotonia, hydrocephalus, and seizures occur less frequently. Radiographically, in comparison to type I, there is better ossification of the vertebrae, ossified fibulae are usually present, and the metacarpals and phalanges are uniformally ossified. Respiratory complications and cervical spine instability often lead to death in the newborn period. However, survival to adulthood has been documented in one case, a woman who subsequently gave birth to an affected child. Inheritance is autosomal dominant, with most cases representing fresh gene mutations. The neck is short, the Atelosteogenesis, Type I short-limb skeletal dysplasia, Am J Med Genet 13:7, 1982. Note the depressed nasal bridge, flexion contractures at knees, and equinovarus position of feet. B, the radiograph reveals lack of calcification of humerus and hypoplasia of much of the skeleton. A, Postmortem photograph of newborn showing limb shortening, a flat face, radial deviation of a low-implanted thumb, and equinovarus with a large gap between toes 1 and 2. B, Note the short limbs, small thorax, bifid distal humeri and rounded iliac bones. F and G, Note, in an adult, the shortening of C2 to C7, flattened bodies of C3 to C6, shortened radius, abnormally shaped carpals, and short metacarpals and phalanges. The designation ` `boomerang dysplasia is generally attributed to Kozlowski et al, who noticed the typical boomerang-like shape of the long tubular bones. Boomerang dysplasia is distinguished from atelosteogenesis on the basis of a more severe defect in mineralization, with complete absence of ossification in some limb bones and vertebrae. Heterozygous mutations associated with the most severe phenotypes almost invariably occur in exons 2-5. These are apparently de novo, but germline or somatic mosaicism has been reported for the milder phenotypes. Histology shows disorganized cartilage of the developing long bone and multinucleated chondrocytes in areas of a hypocellular cartilage matrix. Severe prenatal growth retardation secondary to limb and trunk shortening with sparing of head. Large fontanels, full forehead, hypertelorism, markedly depressed nasal bridge with horizontal groove, hypoplastic nasal septum, micrognathia, short neck with excess skin. Severe micromelia, usually symmetric; abnormal position of the limbs, commonly formed by a single segment; absence of discernible joints; talipes equinovarus. The hands and feet are short and broad and have shortened fingers and toes with poly- or oligodactyly, syndactyly, and hypoplastic nails. Variable delayed calvarial ossification, relative preservation of the thorax, clavicles, sternum and iliac wings with severe delay in mineralization of the vertebrae, pubis, metacarpal/tarsals, and long tubular bones of the arms and legs, boomerang-shaped femora. Sillence D, et al: Atelosteogenesis syndromes: A review, with comments on their pathogenesis, Pediatr Radiol 27:388, 1997. Odent S, et al: Unusual fan shaped ossification in a female fetus with radiological features of boomerang dysplasia, J Med Genet 36:330, 1999. Lu J, et al: Filamin B mutations cause chondrocyte defects in skeletal development, Hum Mol Genet 16:1661, 2007. Note full forehead, hypertelorism, markedly depressed nasal bridge and hypoplastic nasal septum, micrognathia, short neck, and omphalocele (A and B), extremely incurved tibia (C). Histologically, the tibia is the single bone in the middle segment of the lower limb and shows delayed ossification, with a central fibrocartilaginous area with a boomerang shape seen with trichromic stain (D). Primary cilia play an important role in transduction of signals in the hedgehog pathway that is of critical importance in skeletal development. Death from respiratory insufficiency secondary to pulmonary hypoplasia has occurred in all infants in the first few days of life. Short; postaxial polydactyly of hands or feet; syndactyly; metaphyseal irregularities of long bones, with spurs extending longitudinally from medial and lateral segments; underossified phalanges. Short, horizontal ribs; notch-like ossification defects around periphery of vertebral bodies. Small iliac bones with horizontal acetabular roof, triangular ossification defect above lateral aspect of acetabulum. Cardiac defects, including transposition of great vessels, double-outlet left ventricle, double-outlet right ventricle, endocardial cushion defect, and hypoplastic right heart; polycystic kidneys; hypoplasia of penis; defects of cloacal development; imperforate anus. Midline cleft lip; cleft palate; short, flat nose; low-set, small, malformed ears. Both preaxial and postaxial polysyndactyly of hands or feet; brachydactyly; disproportionately short, oval-shaped tibiae; short, rounded metacarpals and metatarsals; premature ossification of proximal epiphyses of humeri, femora, and lateral cuboids; underossified phalanges. Ambiguous genitalia; hypoplasia of epiglottis and larynx; multiple glomerular cysts and focal dilatation of distal tubules of kidney. Short, bowed femora and humeri with cortical thickening of inner midshaft; metaphyses are broad and cupped with osseous spurs projecting laterally in femora, humeri, and phalanges. Narrow and cylindrical with short, horizontal ribs; normally structured vertebral bodies although pedicles of the vertebral arches appear plump; scapulae are square. Mutations in both of these genes have been identified in Jeune thoracic dystrophy, suggesting that they are variants of the same condition. Short, with smooth metaphyseal margins; nonovoid tibia; tibial bones longer than fibular bones; bowed radius and ulna. Narrow and cylindrical with short, horizontal ribs; high clavicles; small scapulae.
Buy 75mg indomethacin with visa
Asymmetric and disproportionate overgrowth of body parts rheumatoid arthritis questions buy discount indomethacin line, normal somatic growth during adolescence and normal final height attainment, tissue overgrowth plateaus after adolescence, macrocephaly. Generalized thickening; epidermal nevi of the flat nonorganoid type; lipomas; asymmetrical subcutaneous fat overgrowth, usually seen over the torso; regional absence of fat; vascular malformations of the venous, capillary, and lymphatic types with a predilection for the thorax and upper abdomen. Hemihypertrophy, scoliosis; kyphosis; hip dislocation; angulation defects of knees; valgus deformities of halluces and feet; macrodactyly; clinodactyly, cerebriform connective tissue nevus involving the soles of the feet, palms, or another part of the body with deep grooves and gyration. Splenomegaly with or without cystic changes; nephromegaly; hydronephrosis; renal calculi and hemangiomas; gastromegaly; colonic polyps; pancreatic lipomatosis; uterine leiomyomatas; hypoplastic uterus, cervical uterine cysts; enlarged ovaries; hypertrophic cardiomyopathy and cardiac conduction defects; pulmonary emphysema, lung cysts and scarring; enlarged thymus. Hyperostosis of skull; abnormal vertebral bodies (asymmetric vertebral body overgrowth, posterior scalloping), segmentation defects, premature degenerative changes; coarse ribs and scapula; abnormal gray-white matter differentiation. A, Note the asymmetric legs, unilateral connective tissue nevus of the foot, long neck, and fat dysregulation. B, Note the facial anomalies, epidermal nevus of the neck, long finger, intra-abdominal lipomas, and locked knee with boney fusion. C, Development and progression of connective tissue nevus in child with Proteus syndrome. E, Severe kyphoscoliosis developing over a 3-year period in a child with Proteus syndrome. Subsequently, more than 54 cases have been reported, and Moog (2009) provides an excellent review. Although motor delay is frequent, the degree of intellectual disability is variable and two thirds of the patients are intellectually normal. Except for skeletal cysts and jaw tumors, the features of this condition are present at birth and are nonprogressive. It is most likely that this disorder is the result of a somatic mutation that is lethal when occurring in the nonmosaic state. Marked developmental delay, intellectual disability (30%), seizures (50%) can be refractory to treatment, spasticity. Hairless fatty tissue nevus of the scalp (nevus psiloliparus) with overlying alopecia, most commonly in the frontotemporal or zygomatic areas, usually unilateral; alopecia without fatty nevus; focal aplastic skin defects; asymmetry of the skull and face; small nodular skin tags on the eyelids or in the area between outer canthus and tragus, which histologically represent fibromas, lipomas, fibrolipomas, or choristomas; unilateral lipomatous involvement of the dermis of the skin covering the face on the same side as the brain defect; ipsilateral skin tags; hard pedunculated outgrowths attached to margin of upper lid made up of connective tissue; unilateral epibulbar or limbal choristoma (dermolipomas and lipodermoids); corneal and scleral abnormalities; ocular and palpebral colobomas; aniridia; microphthalmia; calcification of the eyeglobe; irregular disrupted eyebrows. Intracranial lipomas, most often in the cerebello-pontine angle; spinal lipomas that can extend over the entire spinal cord; arachnoid cysts; unilateral porencephalic cysts; cortical atrophy and calcification of the cerebral cortex overlying the cyst; ventricular dilatation; hemisphere atrophy; defective lamination of the cerebrum; micropolygyri; lipomas in the meninges covering the affected cerebral hemisphere; leptomeningeal angiomatosis. Parazzini C, et al: Encephalocraniocutaneous lipomatosis: Complete neuroradiologic evaluation and follow-up of two cases, Am J Neuroradiol 20:173, 1999. Hauber K, et al: Encephalocraniocutaneous lipomatosis: A case with unilateral odontomas and review of the literature, Eur J Pediatr 162:589, 2003. Svoronos A, et al: Imaging findings in encephalocraniocutaneous lipomatosis, Neurology 77:694, 2011. Multiple enchondromas are found in Ollier disease and in combination with vascular malformations in a condition reported first by Maffucci in 1881. After puberty, gradual ossification of the enchondromas occurs, resulting in solid deformed bones. The disorder can be mild, but it is often severe enough to require multiple surgical procedures and occasionally amputation. These mutations have been identified in primarily enchondromas and vascular affected areas, but also in blood and other tissues in a mosaic state. The phenotypic differences between these three conditions are determined by the proportion of cells carrying the mutations and by their tissue distribution. Demonstration of organic aciduria is an important adjunct to diagnosis, but not a requisite. Variable early bowing of the long bones, with asymmetric retarded growth; enchondromas (40% unilateral), primarily in the hands, feet, and tubular long bones. Vascular malformations, most frequently located in the dermis and subcutaneous fat adjacent to the areas of enchondromatosis, but may occur anywhere; types of vascular malformations are capillary, venous, and especially phlebectasia, which often have a grape-like appearance; thrombosis of the dilated blood vessels with phlebolith formation occurs in 43% of cases. References Maffucci A: Di un caso di encondroma ed angioma multiplo: Contribuzione alla genesi embrionale dei tumor, Movimento Med Chir 3:399, 1881. Superti-Furga A, et al: Enchondromatosis revisited: New classification with molecular basis, Am J Med Genet Part C Semin Med Genet 160C:154, 2012. Note the vascular lesions on both feet (A), the evidence of enchondromas over the outer aspect of the hand (B), and the multiple enchondromas of the hands in a child who had multiple hemangiomata elsewhere (C). Peutz clearly set forth the disease in 1921, and Jeghers and colleagues further established this disease entity in 1949. An intestinal or extraintestinal cancer develops in approximately 40% to 80% of affected patients. The relative risk for cancer is highest for gastrointestinal cancer and breast cancer. Screening of affected patients as well as potentially affected family members should include colonoscopy, an upper gastrointestinal endoscopy plus small bowel examination, breast examination, mammography and pelvic ultrasonography in females older than 20, and careful examination of testicles in males. Vertical bands of epidermal pigment presenting as blue-gray or brownish spots on lips, buccal mucous membrane, perioral area, around the eyes, nostrils, and the perianal area, the digits and elsewhere. Hamartomatous polyps most frequently in small bowel (jejunum and duodenum more frequently than ileum), stomach and colon, and occasionally in nasopharynx, renal pelvis and urinary bladder, biliary tract, and bronchial mucosa; polyps are usually multiple; adenomatous and malignant changes in the polyps as well as in any area of gastrointestinal tract lined by columnar epithelium have been documented. The polyps have mucosa with interdigitating smooth muscle bundles in a characteristic branching tree appearance. Approximately 35% of patients have extraintestinal malignancies, including bronchogenic carcinoma; benign and malignant neoplasms of the thyroid, gallbladder, and biliary tract; breast cancer, usually ductal; pancreatic cancer; malignant tumors of the reproductive tract, including malignant adenoma of the cervix and ovarian and Fallopian mucinous tumors; unique ovarian sex cord tumors with annular tubules (small and benign) that cause heavy menstrual periods and lead to isosexual precocity; and testicular sex cord and Sertoli cell tumors, leading to sexual precocity and gynecomastia. Seventy percent of patients have some gastrointestinal problem by age 20 years, most commonly colicky abdominal pain (60%), intestinal bleeding (25%), or both. Iron deficiency anemia may result from chronic blood loss, and protein-losing enteropathy has been reported. Natural history of complications from polyps in a family may be a predictor of severity for offspring. Routine endoscopy References Hutchinson J: Pigmentation of the lips and mouth, Arch Surg 7:290, 1896. Jeghers H, et al: Generalized intestinal polyposis and melanin spots of the oral mucosa, lips, and digits: A syndrome of diagnostic significance, N Engl J Med 241:993, 1949. Hemminki A, et al: A serine/threonine kinase gene defective in Peutz-Jeghers syndrome, Nature 391:184, 1998. A and B, Spotty pigmentation of lips and buccal mucous membrane in a 4-year-old girl. Subsequently, Dvir and colleagues added Riley-Smith syndrome and suggested that all three of these conditions represent one etiologic entity, which Cohen referred to as BannayanRiley-Ruvalcaba syndrome. Rapamycin has been used in a patient with extensive arteriovenous malformation with good response. At least 10% are caused by intragenic or large deletions, which makes necessary a combined sequencing and dosage testing approach. Birth weight greater than 4 kg and birth length greater than 97th percentile, normal adult stature. Hypotonia, gross motor and speech delay (50%), mild-to-severe mental deficiency (15% to 20%), seizures (25%), autism spectrum disorders. Macrocephaly with ventricles of normal size (mean occipital frontal circumference +4. Hamartomas that are lipomas (75%); vascular malformations, most commonly fast flow arteriovenous malformation (10%); and mixed type (20%). Tan, nonelevated spots on the glans penis and shaft not always present at birth; myopathic process in proximal muscles (60%); cutaneous angiolipomas (50%), encapsulated or diffusely infiltrating; joint hyperextensibility; pectus excavatum; scoliosis (50%). Many affected families have been reported, and the incidence is approximately 1 in 50,000. Of major concern is the potential for brain abscess, cerebral embolism, and hypoxemia secondary to the pulmonary arteriovenous fistulas. Hepatic arteriovenous fistula can cause hepatomegaly, right upper quadrant pain, pulsatile mass, a thrill, or bruit. Left to right shunting through the fistula can lead to high-output congestive heart failure. Specific guidelines have been developed for management of the vascular findings in these patients.
Pimela luzonica (Elemi). Indomethacin.
- How does Elemi work?
- Stomach conditions and coughs.
- What is Elemi?
- Dosing considerations for Elemi.
- Are there safety concerns?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96441

Purchase genuine indomethacin on-line
Evidence that a number of affected individuals have been born to women who experienced events during pregnancy that could cause transient ischemic/hypoxic insults to the fetus suggests that this disorder may be due to any event that interferes with the uterine/fetal circulation arthritis in knee getting worse order indomethacin pills in toronto. The association of seventh cranial nerve palsy with or without sixth cranial nerve palsy but without limb reduction defects may be familial with an autosomal dominant mode of inheritance in some cases. Charles S, et al: Mobius sequence: Further in vivo support for the subclavian artery supply disruption sequence, Am J Med Genet 47:289, 1993. Finally, cytogenetically visible apparently balanced translocations or interstitial deletions involving chromosome 3q2 account for approximately 2% of cases. It is important to distinguish between the types in order to provide counseling to effected individuals and their families relative to reproductive capabilities and menstrual irregularities, including amenorrhea in females with type I. With the exception of infertility in females, the two types are indistinguishable clinically. Therefore, separating the two types can be accomplished only through a combination of molecular testing and careful family history. Inverted inner canthal fold between upper and lower lid, short palpebral fissures with lateral displacement of inner canthi, low nasal bridge and ptosis of eyelids, hypoplasia, fibrosis of the levator palpebrae muscle, strabismus, amblyopia, eyebrows increased in their vertical height and arched. Females with type I have menstrual irregularities or amenorrhea, infertility, and elevated gonadotropin levels. Amblyopia, which occurs in more than 50% of patients, is most frequently associated with asymmetrical ptosis, although it also occurs when the ptosis is bilateral. Although most women with type I have a normal menarche and initially may be fertile, they soon develop ovarian resistance to gonadotropins or true premature ovarian failure. In at least one case, primary ovarian failure has been documented in early childhood. Zlotogora J, Sagi M, Cohen T: the blepharophimosis, ptosis and epicanthus inversus syndrome: Delineation of two types, Am J Hum Genet 35:1020, 1983. Beaconsfield M, et al: Visual development in the blepharophimosis syndrome, Br J Ophthalmol 75:746, 1991. The rounded contour of the "cleft" palate in some of these patients (see illustration) is compatible with this mode of developmental pathology and differs from the usual inverted V shape of most palatal clefts. The focus of management in the newborn period should be treatment of upper airway obstruction and feeding problems. Airway obstruction can lead to hypoxia, cor pulmonale, failure to thrive, and cerebral impairment. Therefore, affected children should be monitored carefully during that period, focusing on the obstruction pathogenesis of the apnea and airway concerns in the condition. In that significant hypoxia may occur without obvious clinical signs of obstruction, serial polysomnography may be helpful over the first month to identify infants at significant risk. Feeding problems requiring nasogastric tube feeding are common and are related in many cases to lower esophageal sphincter hypertonia, failure of lower esophageal sphincter relaxation at deglutition, and esophageal dyskinesis. In 40% of cases, the Robin sequence occurs in otherwise normal individuals, in whom the prognosis is very good if they survive the early period of respiratory obstruction. However, this disorder commonly occurs as one feature in a multiple malformation syndrome of genetic etiology, the most common of which is Stickler syndrome. The fact that accurate diagnosis of a genetic syndrome is often difficult in the newborn period highlights the need for longitudinal follow-up of affected children. Patients who have Robin sequence as one feature of a multiple malformation syndrome require more aggressive airway and feeding management. The Robin sequence may also be a result of early in utero mechanical constraint, with the chin compressed in such a manner as to limit its growth before palatine closure. Baujat G, et al: Oroesophageal motor disorders in Pierre Robin syndrome, J Pediatr Gastroenterol Nutr 32:297, 2001. C, Note the unusual rounded shape to palatal "cleft" in a patient with the Robin sequence compatible with the incomplete closure of the palate having been secondary to the posterior displacement of the tongue. A failure of lip fusion, as shown, may impair the subsequent closure of the palatal shelves, which do not completely fuse until the eighth to ninth week. Other secondary anomalies include defects of tooth development in the area of the cleft lip and incomplete growth of the ala nasi on the side of the cleft. There may be mild ocular hypertelorism, the precise reason for which is undetermined. Tertiary abnormalities can include poor speech and multiple episodes of otitis media as a consequence of palatal incompetence and conductive hearing loss. The highest birth prevalence is in Asians and Native Americans (1 in 500), followed by Europeans (1 in 1000), and the lowest prevalence is in populations of African descent (1 in 2500). The more severe the defect is, the higher the recurrence risk is for future siblings. The following are the general risk figures: unaffected parents with one affected child, 4% for future siblings; unaffected parents with two affected children, 10% for future siblings. As many as 15% of infants surviving the newborn period with cleft lip, with or without cleft palate and 42% of those with cleft palate alone have the defect as part of a broader pattern of altered morphogenesis. One should identify such individuals before using the previously mentioned figures for recurrence risk counseling. In addition, prenatal exposure to valproic acid, maternal smoking and alcohol, and mycophenolate mofetil have been identified as environmental factors associated with cleft lip with or without cleft palate. References Bixler D: Heritability of clefts of the lip and palate, J Prosthet Dent 33:100, 1975. Right, Spontaneously aborted 35-day embryo with hypoplasia of the left lateral nasal swelling and, therefore, a cleft lip. Lower lip pits (80%); hypodontia, missing central and lateral incisors, canines, or bicuspids; cleft lip with or without cleft palate, cleft palate alone, submucous cleft palate, cleft uvula. Mutations in Van der Woude syndrome are References Van der Woude A: Fistula labii inferioris congenita and its association with cleft lip and palate, Am J Hum Genet 6:244, 1954. Janku P, et al: the Van der Woude syndrome in a large kindred: Variability, penetrance, genetic risks, Am J Med Genet 5:117, 1980. Sander A, et al: Evidence for a microdeletion in 1q32-41 involving the gene responsible for Van der Woude syndrome, Hum Mol Genet 3:575, 1994. Oberoi S, Vargervik K: Hypoplasia and hypodontia in Van der Woude syndrome, Cleft Palate Craniofac J 42:459, 2005. Sedano and colleagues subsequently extended these observations and recommended frontonasal dysplasia as a more appropriate designation for this defect. The accompanying illustration sets forth a crude interpretation of the developmental pathogenesis and gradations of the sequence. A number of additional disorders in which frontonasal dysplasia is one feature have been described (see Comment section). Features include extreme microphthalmia, bilateral oblique facial clefts, cleft palate, hypertelorism, wide nasal bridge with hypoplasia of ala nasi, and low-set, posteriorly rotated ears. Features include hypertelorism, wide nasal bridge, short nasal ridge, bifid nasal tip, broad columella, widely separated slit-like nares, long philtrum with prominent vertical ridges, midline notch in upper lip and alveolus. Features include alopecia, a large skull defect, coronal craniosynostosis, hypertelorism, depressed nasal bridge, bifid nasal tip, hypogonadism, callosal body agenesis, and intellectual disability. Several of the affected children were the offspring of consanguineous marriages, raising the possibility of autosomal recessive inheritance. Features include frontonasal dysplasia with optic disk anomalies, basal encephalocele, absent corpus callosum, diabetes insipidus, and pituitary deficiency. Variability from notched broad nasal tip to completely divided nostrils with hypoplasia to absence of the prolabium and premaxilla with a median cleft lip, variable notching of alae nasi, broad nasal root, lack of formation of nasal tip. DeMeyer noted that 8% of his patient population (N = 33) had severe intellectual disability and 12% had impairment of intelligence. The first three of these disorders are due to autosomal recessive mutations in aristaless-like References DeMyer W: the median cleft face syndrome: Differential diagnosis of cranium bifidum occultum, hypertelorism, and median cleft nose, lip, and palate, Neurology [Minn] 17:961, 1967.

Buy cheap indomethacin 75mg online
They show extraordinarily wide polymorphism arthritis neuropathy cheap 75mg indomethacin with amex, but are uniform within an individual. The peptides are derived by proteolytic degradation of endogenous antigens, derived for example from intracellular viruses, by the action of a large multifunctional protease (see Chapters 64 and 65). Some 20 loci affect cytokine levels, signalling pathways in immune cells and non-immunological steps in tissue damage. Selfrecognizing B lymphocytes are likewise eliminated in the bone marrow, although some may survive if the self-antigen concentration is low. Presentation of microbial products to T cells by immature dendritic cells lacking the full complement of co-stimulatory molecules may constitute a negative signal, so inducing tolerance. In addition, T cells contribute to tolerance by sometimes transmitting negative signals. This genetic association is thought to involve interference with the normal immune response to the bacterium Klebsiella. Its persistent inhibition is associated with apoptosis, abnormal immune cell development and delayed cell growth. Tissue incompatibility in transfusion and transplantation As a general rule a recipient will reject a tissue graft from a person who possesses a cell surface antigen absent from the recipient. Relative risk = ad/bc, where a = number of patients with the antigen, b = number of controls with the antigen, c = number of patients without the antigen, d = number of controls without the antigen. A1 and B8 show linkage disequilibrium of association in western Europeans, that is the A1/B8 combination is common. The chance of a random match between unrelated individuals is 1/200 000 so, apart from grafts into privileged sites, successful transplantation generally requires pharmaceutical immunosuppression. The centre pattern is what would be obtained by competitive hybridization of the mixed red and green samples, denatured and hybridized to the same micro-array. Although largely superseded now, the components of the original method are applied in many other techniques. These variant cutting sites provide valuable markers for disease genes that have been exploited in linkage studies (see Chapter 34), though these days, other techniques are generally used. Gel electrophoresis A gel is a three-dimensional mesh with pores of different sizes. They are cast as slabs of agarose or polyacrylamide, with a row of wells at one end for insertion of samples. Alternatively, probes may be linked to molecules that can be detected through non-radioactive means. Diagnostic applications One application involves selection of a restriction endonuclease for which the recognition site corresponds with either the mutant or normal version of the sequence in question. Presence or absence of the mutation can then be determined by comparing fragment sizes. This approach offers an inexpensive and rapid method of diagnosis, but is limited by the need for prior knowledge of mutant sequences. White blood cells are embedded in agarose blocks which are exposed to proteolytic enzymes that digest away cellular materials, leaving the chromosomes intact. This allows examination and separation of very large genes, like dystrophin, and multigenic functional units. Array comparative genome hybridization is a recent method for detecting or locating chromosomal microdeletions. This approach exploits the extraordinary miniaturization and accuracy of inkjet printing and the speed of computer-based analysis. A microarray of dots of cloned oligonucleotides would typically cover the whole genome, with 100 000 oligonucleotides corresponding to sites spaced <30 kb apart throughout. After washing off unbound probes, the fluorescence at both wavelengths is measured and the red: green ratio calculated for each clone. To determine whether a known mutation, or the normal sequence, is present generally involves knowing the sequence for a short stretch around that site. In an individual heterozygous for a single base substitution two different bases would be present there, one on each strand. Heterozygous insertions or deletions produce a complex pattern of superimposed sequences. Not finding a mutation does not necessarily mean no mutation is present, as causative errors may exist in regulatory regions outside the structural gene. It should also be recognized that not all sequence variants disrupt gene function, as would be necessary for it to constitute a pathogenic mutation. Furthermore, deletion or duplication of an entire gene can remain undetected by sequencing. Evidence that a variant is pathogenic might include inference from its likely impact on the gene product; for example, mutations that cause frameshifts are more likely to disrupt function than single base substitutions (see Chapter 25). Demonstration that a mutation is present only in affected individuals and segregates in families, together with disease, is highly suggestive of that mutation being the cause of the disorder, or else is in close linkage with it. Another important clue is recapitulation of the mutant phenotype in an animal model based on the known mutation. In some cases, computer programs can be used to model the effects of a mutation on the functioning of the protein. Direct sequencing is currently not the method of choice for most diagnostic laboratories, when the goal is to identify the presence of a limited repertoire of mutations. Here mutant sites are highly diverse and widely scattered, making it necessary to scan gene sequences in full. Four parallel base-specific reactions are conducted using a mix of all four normal nucleotide precursors, one with a radioactive label, plus a small proportion of one of the four dideoxy-derivatives. If the concentration of the latter is low compared to that of its normal analogue, chain termination occurs randomly at each of the many positions containing that specific base. Each base-specific reaction generates many fragments of different lengths, with variable 3 termini but a common 5 end, corresponding to the primer. Following electrophoresis the gel was dried out and an autoradiographic (or X-ray) film placed in contact with it. After suitable exposure the film was developed to produce a pattern of dark bands. The sequence was then read off by simple inspection of the autoradiograph, providing the 5-to-3 sequence of the new strand complementary to the original template. As they migrate past a window they are excited by a beam of laser light and emit coloured fluorescence. The position of a mutation is then located by comparison of mutant and normal sequences. These adaptations allow sequencing to proceed at a rate of up to a million bases per day. Iterative pyrosequencing Iterative pyrosequencing is a very rapid process that eliminates timeconsuming electrophoresis. Clonal amplification then occurs within each emulsion droplet until 10 million copies of each segment has been produced and immobilized on that bead. Chemiluminescent light is generated with every nucleotide incorporated, according to base pairing rules. Hundreds of thousands of short sequences can be read in parallel, with an overall through-put 10 000 times that of dideoxy-sequencing. There are currently multiple commercial systems that employ various approaches to massively parallel sequencing, making this currently one of the most rapidly advancing areas in genomic technology. Sickle cell status revealed by dot-blot hybridization with a labelled -globin probe HbA/HbA HbA/HbS HbS/HbS HbA primer HbS primer Medical Genetics at a Glance, Third Edition. Analyses can be performed on samples as minute as a single nucleus obtained from a preimplantation embryo, a mouthwash, hair root or other source. Primers are used that flank one specific region of a gene, or alternatively multiple regions such as several exons. An endonuclease is chosen that has a recognition sequence spanning the mutant site (see Chapter 67). Analysis of fragment sizes by agarose gel electrophoresis then indicates the genotype.
Purchase indomethacin toronto
These problems can often be overcome in practice by means of several markers located either side of the disease locus arthritis pain feels like cheap indomethacin 75 mg without a prescription. Linkage analysis is increasingly being replaced by direct analysis, but there are still situations where it is useful. This is due to recombination, the probability of which is known from other studies to be 1%. If necessary, additional polymorphic flanking markers can be used to confirm such deductions and increase the accuracy of analysis. The association may indicate direct involvement of the polymorphic gene in pathogenesis, or only linkage disequilibrium with a nearby disease gene, but in either case, determination of the genotype of an individual can be used to assess the odds of disease (see Chapter 31). If its population prevalence is known this can be used to estimate the absolute risk of disease. Such calculations can be done on a geneby-gene basis, or an individual can be genotyped simultaneously for hundreds of thousands of polymorphisms, yielding a risk profile for a large number of distinct multifactorial conditions. In such cases one must use the situation in an affected child to infer coupling phases in the parents. Those deduced to be at low risk can still develop the condition, while those at high risk may not. There is particular concern that lowrisk individuals may make decisions that have long-term health implications irrespective of genotype, such as to abandon recommended exercise or dieting programmes. This is controversial since such testing may bypass medical professional input to the consumer and important health inferences can be overlooked. X-linkedtraits the same analysis can be done for X-linked traits, tracking the inheritance of the two X chromosomes from a heterozygous female. If the grandparental generation is not available for study, one can infer coupling phase from affected children, with the caveat that one or more might be recombinants. This caveat introduces some uncertainty, and thus decreases analytical power, but it is still better than the Mendelian estimate of 50% based on the equal probability of inheriting one or the other chromosome homologue. Each family must be studied individually, although if there is linkage disequilibrium one particular linked allele can be non-randomly associated with disease (see Chapters 31 and 66). The chorion (syncytiotrophoblast) and bone marrow normally contain sufficient dividing cells for examination, but most tissues require culturing in vitro, with an overall time schedule of about 10 days. Positions of genes along chromosome arms are defined by region number (from the centromere outwards), band, sub-band and sub-sub-band numbers, for example 12q24. High-resolution banding involves fixation before the chromosomes are fully compacted. Preparation of a karyotype A visual karyotype is prepared by arresting dividing cells at metaphase with a spindle inhibitor such as colchicine (see Chapter 16), spreading the cells on a glass slide and staining with Giemsa stain. With chromosomespecific probes it allows rapid diagnosis or exclusion of a diagnosis of trisomy in amniotic fluid cells. In a typical application, a labelled probe is denatured by heating, added to a metaphase chromosome spread on a microscope slide and incubated overnight to permit sequence-specific hybridization. Surplus probe is then washed off and the bound probe located by overlaying Medical Genetics at a Glance, Third Edition. Such probes are created by assembling many copies of the abnormal chromosome using a fluorescence activated chromosome sorter (see Chapter 32). The target can be either a metaphase spread, or an array of tiny samples of oligonucleotides on a glass slide. It is especially valuable in evaluation of individuals with intellectual disability and/or congenital anomalies and is beginning to be used in clinical evaluation of genetic rearrangements in cancer. The two samples are mixed, hybridized competitively to metaphase chromosomes and photographed using a fluorescence microscope. It is possible to create arrays that cover the entire human genome and carry out rapid robotic scans for microdeletions and microduplications. Unbound reporter is washed off and a counterstain applied to reveal the chromosomes. Bound reporter, and hence the site of the gene of interest, is then located by its fluorescence under ultraviolet light. Use of unique sequence probes Microdeletions Submicroscopic deletions can be detected with fluorescent probes directed against one or more unique sequences within the interval suspected to be deleted. Indications for chromosome analysis the following are situations in which cytogenetic investigation is advised: 1 suspected chromosome abnormality; 2 multiple congenital anomalies and/or developmental retardation; 3 disorders of sexual function; 4 undiagnosed intellectual disability; 5 certain malignancies; 6 infertility or multiple miscarriage; 7 stillbirth and neonatal death. Chromosome painting Chromosome painting has now largely been replaced by microarray methods (see Section 13), but may still be useful in some circumstances. Euploidy means that the chromosome number per body cell is an integral multiple of the haploid number, N = 23, aneuploidy that it is other than an integral multiple. Diploidy describes the normal situation, a typical body cell in humans having 2N = 46 chromosomes. Chromosomal abnormalities are present in at least 10% of spermatozoa and 25% of oocytes. Approximately 50% of spontaneous first trimester miscarriages have a chromosome abnormality, including a high proportion of Trisomy 13 (T13) and T18 fetuses. The birth frequency of this class increases with maternal age, especially after 35 years of age. Translocations that lead to partial trisomy are associated with milder manifestation and longer survival. Low nasal root, ears small Eyes slanting upwards, with marked epicanthic folds; cataracts, squint and nystagmus (involuntary eye movements). There is usually significant intellectual delay, with specific deficits in speech and auditory short-term memory. Life expectancy is reduced and there are Alzheimer-like features in half those over 40 years of age. There is risk of obesity, sleep apnoea and skeletal problems, including dislocation of cervical vertebrae. Karyotype formulae indicate the total number of chromosomes, together with the sex chromosome constitution. Bodyform Phenotype is basically male, tall with elongated lower legs and forearms, but with a feminine body shape and low muscle mass. Fertility Small, soft testes (<10 mL, 2 cm); most are sterile or produce few sperm, as a result of atrophy of the seminiferous tubules. Pubic, axillary and chest hair are sparse and daily facial shaving is rarely necessary. Otherfeatures There may be scoliosis, emphysema, varicose veins and leg ulcers, diabetes mellitus in 8% and thyroid problems are common. Mental deficiency and physical abnormality increase with the number of supernumerary X chromosomes. Management Klinefelter syndrome presents in childhood with clumsiness, learning difficulties and poor verbal skills. Testosterone therapy by long-term implants should be initiated at the beginning of puberty. Turner syndrome, X chromosome monosomy Genetics Karyotype 45,X; body cells abnormal for females in containing no Barr bodies. Mature height averages 145 cm (4 ft 9 in; 20 cm below average); shield-shaped chest with widely spaced nipples. Headandface Heart-shaped face with micrognathia and low posterior hairline; excess skin forms a web between neck and shoulders; high arched palate with overcrowding of teeth.
Order cheap indomethacin on-line
A and B: this bone marrow biopsy demonstrates extensive involvement by follicular lymphoma bauer black arthritis relief gloves indomethacin 25mg on-line, with the multiple paratrabecular lymphoid aggregates closely hugging bony trabeculae. C: Closer inspection reveals that the lymphoid infiltrate consists primarily of small lymphocytes with coarse chromatin pattern and irregular nuclear contours. A: Medium-power view of biopsy from a case of follicular lymphoma show the characteristic paratrabecular aggregates. Nuclear morphology of chronic lymphocytic leukemia (A), follicular lymphoma (B), and large B-cell lymphoma (C) lymphocytes in biopsy. Although lymphomas should not be classified based on their appearance in bone marrow, this composite figure highlights the differences in nuclear morphology among these three different B-cell lymphoproliferative disorders. The ideograms of chromosomes 14 and 18 and of the respective derivative chromosomes are to the left in color. Most cases of follicular lymphoma have a t(14;18) reciprocal chromosomal translocation. This protein is not detected in high amounts in benign, reactive germinal center cells. Diagrams in the left panels show the chromosomal localization of breakpoints in the two component genes. Malignant B-cell proliferations are clonal, a feature that helps distinguish them from reactive polyclonal proliferations. This lymph node shows the homogenous, fish-flesh appearance of lymphoma totally replacing the entire lymph node. An aspirate smear from the same case shows a pleomorphic population of highly atypical, large lymphoid cells (right lower panel). Low- (top), medium- (middle), and high- (bottom) power views demonstrate numerous large, poorly circumscribed, mid-trabecular aggregates made up of a heterogeneous population of large, atypical lymphoid cells with vesicular nuclei admixed with small mature lymphocytes. This lymph node shows marked sclerotic thickening of its capsule, with complete effacement of the architecture by extensive, noncohesive sheets of monotonous, large cells. A high mitotic rate with tripolar mitotic figure (white triangle) and multilobed nuclei (small black triangle) is seen, as well as numerous apoptotic bodies (small arrows in bottom right smear). This autopsy specimen with transversely cut lungs and mediastinum at the level of the aortic arch demonstrates a large, fibrotic, infiltrative mass encasing the entire aortic arch and extending into the adjacent lung. A biopsy from the base of the tongue shows necrosis and extensive infiltration of tissue with atypical large lymphoid cells composed of immunoblasts with large, single, central nucleoli (white arrows) and centroblasts with multiple smaller nucleoli situated near the nuclear membrane (black arrows). These blood smears show circulating, large nucleolated lymphoid cells, some with clefted nuclear contours. For comparison, mature small lymphocytes are present near the lower left corners of both figures. C: the bone marrow biopsy is totally replaced by sheets of poorly preserved, malignant cells that display extensive crush artifact. These "double-hit" lymphomas, as well as diffuse large B-cell lymphoma, when confined to blood and marrow at the time of diagnosis, are easily misdiagnosed as precursor B-cell lymphoblastic leukemia/lymphoma. This cervical lymph node specimen showed total effacement by a proliferation of large atypical lymphoid cells with prominent nucleoli and in association with tangible body macrophages. An autopsy lung from a patient with lymphomatoid granulomatosis reveals extensive involvement with multiple necrotizing lesions. B: Lung biopsy showing large lymphoid cells restricted to blood vessels and displaying a vasculitic pattern of involvement, with fibrinoid necrosis and angioinvasion. The lymphoma cells extend through the walls of the vessels but do not invade brain parenchyma. A and B: A supraclavicular lymph node biopsy shows dense fibrous tissue and extensive coagulative tumor necrosis intimately associated with sheets of large, malignant lymphoid cells. C: the malignant cells have giant, eosinophilic staining nucleoli (black arrow) and mitotic figures (white arrow). A: the biopsy of an abdominal mass shows, at low power, the classic "starry sky" appearance of rapidly proliferating malignant cells intermixed with larger histiocytes actively engulfing tumor debris. B: Higher power shows intermediatesized cells that are uniform in size and shape, with multiple small basophilic nucleoli. This bone marrow biopsy demonstrates total replacement by rapidly growing tumor cells, some of which exhibit apoptosis admixed with "starry-sky" histiocytes engulfing tumor debris. This blood smear reveals L3-type lymphoblasts with multiple nucleoli and basophilic staining, vacuolated cytoplasm. The ideograms of chromosomes 8, 14, and the respective derivative chromosomes are to the left in color; the corresponding G-banded chromosome pairs are to the right. The tumor contains sheets of atypical plasmacytoid cells with a high mitotic rate. Gross lymph node specimens have been cut to show characteristic homogenous pattern ("fish flesh" appearance) of a lymph node diffusely involved by chronic lymphocytic leukemia/small lymphocytic lymphoma (A), as compared with the nodular, heterogeneous appearance of a lymph node involved with metastatic cancer (B). Frequent smudged or "basket" cells, which are nuclear remnants of damaged cells, can be seen. B: Mantle cell lymphoma has small lymphocytes, condensed chromatin, and scant cytoplasm. C: Hairy cell leukemia has lymphopenia, ample cytoplasm with a central nucleus, and cytoplasmic projections. D: Splenic marginal zone lymphoma has lymphocytosis and irregular, eccentric nuclei. An internal mammary node is diffusely effaced by a proliferation of small, mature lymphocytes with high N:C ratios and condensed chromatin. Pale staining proliferation centers are composed of prolymphocytes and immunoblasts-larger cells with vesicular nuclei and nucleoli. The lymph node is diffusely replaced by small mature lymphocytes with high N:C ratios and condensed chromatin. Chronic lymphocytic leukemia/small lymphocytic lymphoma; diffuse marrow involvement. Diffuse infiltration involves complete effacement of bone marrow architecture by sheets of lymphoma cells. D: Deletion of the p53 gene (red) at 17p13 as compared with a centromere 17 control (green). In this gross specimen, the spleen is homogeneously enlarged by diffuse infiltration of the red pulp. For comparison, a normal small lymphocyte is present in the upper left corner of the figure. Multiple small (1- to 3-mm) discrete nodules of expanded white are diffusely distributed throughout the splenic parenchyma. The spleen is diffusely infiltrated by macroscopically visible nodular expansions of white pulp. Perivascular nodules made up of monotonous-appearing, well-spaced, medium-sized lymphoid cells are shown. The typical cells in this disease are mature-appearing, medium-sized lymphocytes with ample cytoplasm that forms villous projections. A: Marrow biopsy shows a mostly sinusoidal pattern of marrow involvement by lymphoma. B and C: Highpower views show small-sized lymphoma nuclei (compared with the larger endothelial nucleus; arrow in B), which possess a condensed chromatin pattern and regular nuclear contours. Clinical photograph of eye showing fleshy "salmon-color patch" of the superficial ocular surface. This gastric biopsy demonstrates extensive involvement by well-spaced, medium-sized lymphoid cells that invade and destroy glands to form lymphoepithelial lesions (arrow). Extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue lymphoma. Uniformly, well-spaced, medium-sized lymphoid cells invade and destroy glands to form lymphoepithelial lesions (bottom right image). A lymph node and subjacent parotid gland demonstrate a vaguely nodular proliferation (A and B) of well-spaced monocytoid lymphocytes invading germinal centers (C) and glandular structures to form lymphoepithelial lesions (D). Mucosa-associated lymphoid tissue lymphoma; lymph node involvement by extranodal marginal zone B-cell lymphoma. This lymph node is completely effaced by diffuse sheets of monotonous-appearing plasma cells, some showing intranuclear inclusions of monoclonal immunoglobulin in the form of Dutcher bodies (arrows).
