

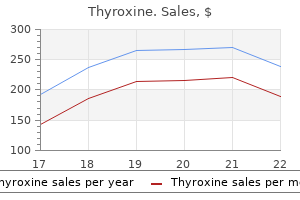
Purchase discount thyroxine online
The molecular weight (about 293) is low enough that excretion into breast milk should be expected 2d6 medications purchase cheapest thyroxine. Because of the long terminal elimination half-life (approximately 42 hours or longer) in adults and the unknown amount of oxaprozin that is excreted into milk, any of these choices is probably preferable. Although a number of studies have reported an association with various types of congenital defects including the study below, other studies with this class of drugs have not found such associations. Maternal denial of exposure and the concurrent exposure to other toxic drugs and substances. Continuous use during gestation or close to delivery might result in neonatal withdrawal or a dose-related syndrome similar to that observed with diazepam. The drug, both free and conjugated forms, crosses the placenta achieving average cord:maternal serum ratios of 0. Large variations between patients for placental transfer have been observed (13). Passage of oxazepam is slower than that of diazepam, but the clinical significance of this is unknown (4). Two reports have suggested that the use of oxazepam in preeclampsia would be safer for the newborn infant than diazepam (5,6). A 1989 report described characteristic dysmorphic features, growth restriction, and central nervous system defects in eight infants exposed either to oxazepam (75 mg/day) or diazepam (30 mg/day) (7) (see Diazepam for a detailed description of the infants). The authors concluded that the clinical characteristics observed in the infants probably represented a teratogenic syndrome related to benzodiazepines (7). Oxazepam, an active metabolite of diazepam, was detected in the urine of an infant exposed to high doses of diazepam during lactation (8). The infant was lethargic and demonstrated an electroencephalographic pattern compatible with sedative medication (see Diazepam). Mild facial defects have been observed with oxcarbazepine and, a closely related agent carbamazepine has been associated with neural tube defects and minor craniofacial malformations (see Carbamazepine). Until this information is available, the safest course is to give folic acid supplementation with oxcarbazepine, as is done with other antiepileptic agents. In addition, metabolism of oxcarbazepine does not result in epoxide metabolites (13). Because these intermediate arene oxide metabolites have been associated with teratogenicity (see Carbamazepine, Phenytoin, and Valproic Acid), this may indicate a lower risk of teratogenicity with oxcarbazepine compared with the other agents. The chemical structure of oxcarbazepine is closely related to that of another anticonvulsant, carbamazepine. In pregnant rats, the administration of oral oxcarbazepine during organogenesis, at doses approximately 1. Embryo and fetal death but no birth defects were observed in rabbits given oral doses of the active metabolite (1. In a strain of mice susceptible to the teratogenic effects of carbamazepine, the highest tolerable oral dose of oxcarbazepine (1100 mg/kg/day) produced a lower incidence of teratogenicity (8% vs. Concurrent treatment with phenobarbital did not increase the incidence of congenital malformations over that observed with oxcarbazepine alone (5). In agreement with the molecular weight (about 252) of the parent drug, oxcarbazepine and its metabolite have been found in the fetus (2,6). The healthy 3700-g female infant had mild facial dysmorphism with a discrete epicanthus and a broad nasal bridge. Her development at 13 months of age was normal without signs of mental retardation or neurologic deficit. No placental metabolism of carbamazepine was observed in the perfusion system (2). The author of a 1994 report very briefly reviewed the pregnancy outcomes of 27 women treated with oxcarbazepine (7). A 1996 review discussed the effects of oxcarbazepine and other anticonvulsants (1). Compared with carbamazepine, oxcarbazepine causes less induction of the cytochrome P450 enzyme system. The Lamotrigine Pregnancy Registry, an ongoing project conducted by the manufacturer, was first published in January 1997 (8). There were three exposures in the 2nd/3rd trimesters resulting in live births without defects (8). A 2006 review of prophylactic therapy of bipolar disorder briefly described the effects in pregnancy and breastfeeding of a number of drugs, including oxcarbazepine (9). Although the limited data prevented conclusions on the relative safety of the drugs, the author did state that each case needed to be considered separately (9). Nursing infants should be monitored for poor suckling, vomiting, and sedation (10). However, because the American Academy of Pediatrics classifies carbamazepine as compatible with breastfeeding (see Carbamazepine), oxcarbazepine can probably be similarly classified. A review of its pharmacology and therapeutic potential in epilepsy, trigeminal neuralgia and affective disorders. Pharmacokinetics of oxcarbazepine and 10-hydroxy-carbazepine in the newborn child of an oxcarbazepine-treated mother. Prophylactic treatment of bipolar disorder in pregnancy and breastfeeding: focus on emerging mood stabilizers. Moreover, there is no evidence suggesting that similar topical imidazole derivative antifungal agents cause embryofetal harm, and there is no reason to believe that oxiconazole would be different. It is in the same antifungal class of imidazole derivatives as butoconazole, clotrimazole, econazole, ketoconazole, miconazole, sertaconazole, sulconazole, and tioconazole. Oxiconazole is indicated for the treatment of tinea pedis, tinea cruris, and tinea corporis due to Trichophyton rubrum, T. The cream also is indicated for the treatment of tinea (pityriasis) versicolor due to Malassezia furfur. Information regarding the metabolism, plasma protein binding, and elimination half-life has not been located. Reproduction studies with orally administered oxiconazole have been conducted in pregnant mice, rats, and rabbits. Studies evaluating the carcinogenic potential of oxiconazole have not been conducted by the manufacturer. At higher doses, there was a decrease in fertility parameters in both females and males, decreased number of sperm in vaginal smears, extended estrous cycle, and a decrease in mating frequency (1). The molecular weight (about 492) is low enough for passage, but the minimal systemic bioavailability suggests that exposure of the embryo or fetus is clinically insignificant. The molecular weight (about 492) is low enough for excretion into breast milk, but the minimal systemic bioavailability suggests that exposure of the nursing infant would be clinically insignificant. The drug is a tertiary amine that is a direct inhibitor of smooth muscle spasm of the urinary tract. It also possesses antimuscarinic activity (about one-fifth of the anticholinergic activity of atropine). Oxybutynin is used for the relief of symptoms associated with bladder instability and nocturnal enuresis. It is in the same subclass as darifenacin, flavoxate, solifenacin, tolterodine, and trospium. No fetal harm was noted in female rats dosed orally with oxybutynin, 20160 mg/kg/day (maximum recommended human dose is 20 mg/day or less than 0. The molecular weight (about 358 for the free base) is low enough that exposure of the embryofetus should be expected. A 2001 case report described a paraplegic woman who had used baclofen 80 mg/day (the maximum recommended daily oral dose), oxybutynin 9 mg/day, and trimethoprim 100 mg/day throughout an uneventful pregnancy (5). The fetus developed tachycardia at an unspecified gestational age and an apparently healthy female infant was delivered (birth weight not given). At 7 days of age, the infant was admitted to the hospital for generalized seizures, but the mother had noted abnormal movements 2 days after birth. Because seizures had been reported in adults after abrupt withdrawal of baclofen, a dose of 1 mg/kg divided into four daily doses was started and the seizures stopped 30 minutes after the first dose.
Proven thyroxine 25 mcg
However shakira medicine purchase thyroxine 125 mcg without a prescription, immunoglobulins, such as IgG, readily pass into milk during the colostral phase (first 48 hours) and, because pertuzumab has a long half-life (18 days), doses received late in pregnancy will be excreted into colostrum and possibly into early milk. Moreover, the drug is given in combination with trastuzumab and docetaxel, so a nursing infant might be exposed to three antineoplastics. Consequently, the safest course for the infant is to not breastfeed if the mother is receiving this therapy. An active alkaloid in peyote is mescaline, an agent that causes developmental toxicity in animals. The use of peyote for religious rituals by certain Indian populations involves low doses and does not appear, based on limited data, to cause developmental toxicity. If true, this would be consistent with the principle that developmental toxicity is dose related. However, the apparent low risk of developmental toxicity in Native American populations using carefully monitored amounts of peyote probably is not transferable to the recreational use of peyote, and certainly not to the use of mescaline. Although the recreational use of peyote or mescaline in pregnancy has not been studied, the presumed higher doses might represent a risk to the embryo or fetus. In addition, the lifestyle of recreational users and the probable use of other hallucinogenic or abuse drugs might not be conducive to a healthy pregnancy. Therefore, such use should be discouraged in pregnancy and is classified as contraindicated. Other uses include orally for the treatment of fever, rheumatism, and paralysis, and topically for treating fractures, wounds, and snakebite (1). The active ingredient in peyote is mescaline, a chemical structurally related to amphetamines. Congenital malformations of the brain, spinal cord, liver, and other viscera were increased. The incidence of defects, however, was not dose-related as the lowest dose caused the greatest number of defects (28%), whereas the highest dose caused the fewest (9%). Resorptions (7%25%), dead fetuses (5%10%), and runts (5%12%) were dose related (2). Oral doses of 16 and 32 mg/kg on the 7th through the 10th day of gestation resulted in dose-dependent increases in resorptions and corresponding decreases in litter size. There also was a doserelated increased delay in the ossification of the skull, sternum, and metatarsals. A similar study in mice, using only labeled mescaline given intraperitoneally, found that the drug crossed the placenta and concentrated in fetal organs with the lowest concentrations in fetal brain (5). Although one author stated that mescaline is known to cross the human placenta (6), such studies have not been located. The molecular weight (about 211) is low enough that exposure of the embryo and fetus should be expected. Passage across the human placenta should be limited by the ionization at physiologic pH and the low solubility of unionized mescaline found in the above monkey study. A brief 1970 review summarized the history, dosage, effects, and adverse effects of mescaline (8). Peyote has been used by some Mexican and American Indian tribes for hundreds of years and still is being used legally for religious rituals by the Native American Church. The author thought that mescaline, based on dosage, was the least potent of the commonly used hallucinogens (other hallucinogens not specified). The author also thought that mescaline might be teratogenic based on animal studies (8). A 2001 review provided more information on the legal use of peyote and its pharmacology (6). When peyote is used according to recommendations of the Native American Church, the risk for developmental toxicity was not thought to be increased. No increase in malformations was observed in Huichol Indians, a tribe that has used peyote as part of its religious rituals for centuries. However, based on the 1967 animal study cited above, the author concluded that risk of human developmental toxicity was associated with the recreational use of peyote and/or mescaline in which much higher doses than those in religious rituals are used. In addition, mescaline obtained on the street is often mislabeled and may not contain any mescaline at all. The molecular weight of mescaline (about 211) is low enough that excretion into breast milk should be expected. However, the use of low doses of peyote by adults in religious rituals often is associated with adverse effects, such as nausea, vomiting, and sympathomimetic effects (mydriasis, mild tachycardia, mild hypertension, diaphoresis, ataxia, and hyperreflexia) (6). Higher maternal doses that may be associated with recreational use of peyote, and certainly with mescaline, could cause these effects in a nursing infant. Thus, the recreational use of peyote or mescaline should be considered contraindicated. Congenital malformations induced by mescaline, lysergic acid diethylamide, and bromolysergic acid in the hamster. A brief 2008 review of urinary tract infections in pregnancy also cited the above data (2). In a surveillance study of Michigan Medicaid recipients involving 229,101 completed pregnancies conducted between 1985 and 1992, 496 newborns had been exposed to phenazopyridine during the 1st trimester (F. No anomalies were observed in four other defect categories (spina bifida, polydactyly, limb reduction defects, and hypospadias) for which specific data were available. The molecular weight (about 214 for the free base) suggests that the drug will cross to the embryofetus. In a large 1985 study involving 6509 women with live births, 217 were exposed to phenazopyridine in the 1st trimester (3). The molecular weight (about 214 for the free base) suggests that the drug will be excreted into breast milk. Although the drug does not appear to cause structural anomalies, its use in pregnancy has been associated with functional/neurobehavioral defects. Qualitative analysis of the urine from two newborns revealed phencyclidine levels of 75 ng/mL or greater up to 3 days after birth (2). However, the specificity of the chemical screening methods used in this latter report has been questioned (7). However, case reports involving four newborns indicate that the use of this agent may result in long-term damage (2,9,10): Depressed at birth, jittery, hypertonic, poor feeding (2) (two infants) Irritable, poor feeding and sucking reflex (9) (one infant) Triangular-shaped face with pointed chin, narrow mandibular angle, antimongoloid slanted eyes, poor head control, nystagmus, inability to track visually, respiratory distress, hypertonic, jitteriness (10) (one infant) Irritability, jitteriness, hypertonicity, and poor feeding were common features in the affected infants. In three of the neonates, most of the symptoms had persisted at the time of the report. High levels of the drug caused progressive degeneration and death of the neurons, whereas sublethal concentrations inhibited axonal outgrowth. The abnormality was significantly more than that observed in control infants and those exposed in utero to opiates, sedative/stimulants, or the combination of pentazocine and tripelennamine. The American Academy of Pediatrics considers the drug to be contraindicated during breastfeeding (15). Phencyclidine use among youth: history, epidemiology, and acute and chronic intoxication. Degenerative and axon outgrowth-altering effects of phencyclidine in human fetal cerebral cortical cells. It is indicated for the management of exogenous obesity as a shortterm adjunct (a few weeks) in a regimen of weight reduction based on caloric restriction. The molecular weight (about 191 for the free base) is low enough, but its hydrophilic nature should limit the exposure of the embryofetus. The molecular weight (191 for the free base) is low enough for excretion into breast milk. Milk is slightly acidic compared with plasma and ion trapping could result in milk:plasma ratios >1. Other agents in this subclass are iproniazid, isocarboxazid, nialamide, and tranylcypromine. Plasma protein binding was not reported by the manufacturer, but the mean elimination half-life after a single 30-mg dose was 11. In reproduction studies with mice at doses above the maximum recommended human dose, a significant decrease in the number of viable offspring per mouse was observed (1). The molecular weight of the sulfate form (about 234) and elimination half-life suggest that exposure of the embryofetus should be expected. A 1995 report described a 25-year-old woman who had been treated for depression with phenelzine (45 mg/day) for 6 years before becoming pregnant (3). Because labor failed to progress, a cesarean section was performed to give birth to a healthy male infant with Apgar scores of 9 at 1 and 5 minutes (3).
Order genuine thyroxine line
Nevertheless treatment 32 for bad breath buy thyroxine on line amex, until human pregnancy data are available, the safest course is to avoid solifenacin during pregnancy, especially in the 1st trimester. If inadvertent exposure in pregnancy does occur, the embryo fetal risks probably are low. Solifenacin is indicated for the treatment of overactive bladder with symptoms of urge urinary incontinence, urgency, and urinary frequency. It is in the same subclass as darifenacin, flavoxate, oxybutynin, and tolterodine, and trospium. One of the metabolites is active, but the concentration is low and is not thought to contribute significantly to clinical activity. When this dose was continued in pregnancy and during lactation, reduced peripartum and postnatal survival, reductions in body weight gain, and delayed physical development (eye opening and vaginal patency) were observed. Solifenacin was not carcinogenic in mice and assays for mutagenicity were negative. The molecular weight (about 363 for the free base) and prolonged plasma elimination half-life suggest that the drug and/or its metabolites will cross to the embryofetus. The molecular weight (about 363 for the free base) and prolonged plasma elimination half-life suggest that the drug and/or its metabolites will be excreted into breast milk. Although neonates are particularly sensitive to anticholinergics, two other agents in the anticholinergic class are classified by the American Academy of Pediatrics as compatible with breastfeeding (see Atropine and Scopolamine). The effect of solifenacin exposure in a nursing infant is unknown, but the risk of toxicity probably is low. It should be noted, though, that sorafenib inhibits angiogenesis, a critical component of embryonic and fetal development. The manufacturer recommends that adequate contraception should be used during therapy and for at 2 weeks after completing therapy (1). However, renal cell carcinoma can be fatal, so if a woman requires sorafenib and informed consent is obtained, treatment should not be withheld because of pregnancy. Sorafenib is indicated for the treatment of patients with advanced renal cell carcinoma. After hepatic metabolism, at least eight metabolites have been identified, one of which has activity similar to the parent compound. The drug was clastogenic, in the presence of metabolic activation, in one test but was not mutagenic or clastogenic in other tests. An intermediate in the manufacturing process that also is present in the final product (<0. Specific studies for fertility impairment have not been conducted, but sorafenib does adversely affect male and female reproductive organs. These effects, more pronounced in rats than in mice or dogs, included testicular atrophy and degeneration, oligospermia, degeneration of epididymis, prostate, and seminal vesicles, central necrosis of the corpora lutea, and arrested follicular development at doses that were about 0. The molecular weight (433 for the free acid) and the long elimination half-life suggest that the drug will cross to the embryo and/or fetus. The molecular weight (433 for the free acid) and the long elimination half-life suggest that the drug will be excreted into breast milk. The effects of this exposure on a nursing infant are unknown, but severe toxicity may occur. In adults, handfoot skin reaction and rash were the most common adverse events, but diarrhea, hemorrhage, and hypertension were also common. Several reviews have examined the use of -adrenergic blockers in human pregnancy, concluding that these agents are relatively safe for the fetus (14). Newborns exposed near delivery should be closely observed during the first 2448 hours for signs and symptoms of blockade. Long-term effects of in utero exposure to this class of drugs have not been studied but warrant evaluation. In a 1996 study, the embryotoxicity of d-sotalol in rats was shown to be related to gestational age and resulted from dose-dependent bradycardia in in vitro (rat embryo culture) and in vivo (pregnant rat) experiments (7). In embryo cultures, the minimum effective concentration (15 mcg/mL) for causing significant bradycardia was about five times the human therapeutic plasma concentration (3 mcg/mL) achieved in pregnant women after a 400-mg oral dose. In nine pregnant rats, a single oral dose (1000 mg/kg) was administered on gestational day 13. No malformations were observed, but a resorption rate of almost 14% was noted (7). No data were provided on the time interval between the last sotalol dose and the collection of plasma samples (9). In a second study, eight women scheduled for elective cesarean section were given 80 mg of the drug orally 3 hours before the procedure (10). A 1990 report described the use of sotalol, 80 mg twice daily, in one woman throughout gestation (11). These results suggested that sotalol accumulated in the amniotic fluid, but not in the fetus. In addition, there was no correlation between the effectiveness of sotalol therapy and maternal blood levels (13). Sotalol was used in 12 pregnant women for the treatment of hypertension (details above) (9). No fetal adverse effects attributable to the drug were observed, but bradycardia (90110 beats/minute), lasting up to 24 hours, was discovered in five of the six newborns with continuous heart rate monitoring (9). Therapy was eventually changed to amiodarone plus digoxin with return of a normal fetal heart rate and resolution of the fetal edema. No adverse effects of drug exposure, including bradycardia, were noted in the newborn, which was growing normally at 1 year of age (11). Two fetuses did not respond and two severely hydropic fetuses died 1 and 10 days, respectively, after starting sotalol. Of the surviving infants, 11 were doing well and 1 had cerebral atrophy, hypotonia, and developmental delay thought to be due to the arrhythmia and a thromboembolism (15). A 2000 retrospective study evaluated the use of sotalol for fetal tachycardia in 21 pregnant women (16). Twenty paired samples of breast milk and maternal blood were obtained from 5 of the 12 women treated during and after pregnancy with sotalol for hypertension (dosage detailed above) (9). Specific details of dosage and the timing of sample collection in relationship to the last dose were not given. No -blockade effects were observed in the five nursing infants, including the one infant who had bradycardia at birth. The mother of this infant produced the highest concentrations of sotalol in milk (20. A woman treated throughout gestation with sotalol (80 mg twice daily) and flecainide was continued on these drugs during the postpartum period (11). Simultaneous milk and plasma samples were drawn 3 hours after the second dose of the day on the 5th and 7th postpartum days. The study was repeated on the 105th postpartum day, yielding prefeeding milk and serum concentrations 2. No adverse effects were observed in the infant, who continued to develop normally throughout the study period (12). Although symptoms of -blockade, such as bradycardia and hypotension, were not observed in the nursing infants described above, these effects have been noted with other -adrenergic blockers (see also Acebutolol, Atenolol, and Nadolol) and may occur with sotalol. Long-term effects of exposure to blockers from milk have not been studied but warrant evaluation. The American Academy of Pediatrics classifies sotalol as compatible with breastfeeding (17). Teratogenic potential of almokalant, dofetilide, and d-sotalol: drugs with potassium channel blocking activity. Coadministration of flecainide acetate and sotalol during pregnancy: lack of teratogenic effects, passage across the placenta, and excretion in human breast milk. The animal reproduction is not relevant because oral doses were used and the drug is given topically in humans. Moreover, topical use of the drug suspension does not produce quantifiable amounts of spinosad in human plasma. Although the suspension contains an unspecified amount of benzyl alcohol, it is doubtful if sufficient amounts of the additive would be absorbed to cause embryofetal toxicity.
Buy 25 mcg thyroxine with mastercard
However medicine upset stomach cheap thyroxine 50 mcg, growth and development were normal in 13 infants (one set of twins) examined for 6 months to 10 years (12,15,16,1821). Severe oligospermia has been described in a 22-year-old male receiving sequential chemotherapy of cyclophosphamide, methotrexate, and mercaptopurine for leukemia (23). After treatment was stopped, the sperm count returned to normal and the patient fathered a healthy female child. Ovarian function in women exposed to mercaptopurine does not seem to be affected adversely (2529). An investigator noted in 1980 that long-term analysis of human reproduction following mercaptopurine therapy had not been reported (30). However, a brief 1979 correspondence described the reproductive performance of 314 women after treatment of gestational trophoblastic tumors, 159 of whom had conceived with a total of 218 pregnancies (28). Excluding the 17 women still pregnant at the time of the report, 38 (79%) of 48 women, exposed to mercaptopurine as part of their therapy, delivered live, term infants. A more detailed report of these and additional patients was published in 1984 (31). This latter study and another published in 1988 (32), both involving women treated for gestational trophoblastic neoplasms, are discussed below. In 436 long-term survivors treated with chemotherapy between 1958 and 1978, 95 (22%) received mercaptopurine as part of their treatment regimens (31). Of the 95 women, 33 (35%) had at least one live birth (numbers given in parentheses refer to mean/maximum mercaptopurine dose in grams) (5. A 1988 report described the reproductive results of 265 women who had been treated from 1959 to 1980 for gestational trophoblastic disease (32). Single-agent chemotherapy was administered to 91 women, including 26 cases in which mercaptopurine was the only agent used, whereas sequential (single agent) and combination therapy was administered to 67 and 107 women, respectively. Of the total group, 241 were exposed to pregnancy and 205 (85%) of these women conceived, with a total of 355 pregnancies. A total of 303 (4 sets of twins) liveborn infants resulted from the 355 pregnancies, 3 of whom had congenital malformations: anencephaly, hydrocephalus, and congenital heart disease (1 in each case). Cytogenetic studies were conducted on the peripheral lymphocytes of 94 children and no significant chromosomal abnormalities were noted. Moreover, follow-up of the children, with >80% of the group >5 years of age (the oldest was 25 years), revealed normal development. The reproductive histories and pregnancy outcomes of the treated women were comparable to those of the normal population (32). A 2012 case described a 27-year-old woman with ulcerative colitis who was treated throughout pregnancy with allopurinol 100 mg/day, mercaptopurine 25 mg/day, and mesalazine 4 g/day (33). An elective cesarean section was performed at 39 weeks to give birth to a healthy 3550-g infant (sex not specified) with Apgar score of 9, 10, and 10 at 1, 5, and 10 minutes, respectively. The molecular weight (about 170) suggests that the drug will be excreted into milk. Pregnancy outcomes after successful chemotherapy for, choriocarcinoma and invasive mole: long-term follow-up. Although the limited human pregnancy experience does not allow a full assessment of the embryofetal risk, another carbapenem antibiotic is considered safe to use during the perinatal period. Meropenem is indicated as single-agent therapy for the treatment of complicated skin and skin structure infections (adult patients and pediatric patients 3 months only), complicated intra-abdominal infections (adult patients and pediatric patients 3 months only), and bacterial meningitis (pediatric patients 3 months only). Plasma protein binding is minimal (about 2%) and the elimination half-life is about 1 hour (1). Placental passage in humans has apparently not been studied, but the molecular weight (about 384) suggests that the drug will cross to the embryo fetus. Moreover, the drug is distributed into many human tissues, including the endometrium, fallopian tubes, and ovaries (1). The woman went into premature labor 1 month after completion of the therapy and delivered a baby without evidence of infection or abnormalities (no other details were provided) (2). She responded well to the therapy and was then given amoxicillin (750 mg/day) for 4 weeks. A 2012 case report described the use of meropenem (3 g/day) for 7 days starting on postpartum day 6 by a mother exclusively breastfeeding her infant (4). In another case, a mother breastfed her infant until the 4th postpartum month (5). At 2 months, the mother was treated with a 2-week course of meropenem and tobramycin (doses not specified) for a cystic fibrosis exacerbation. In the above cases, the absence of toxic effects in the nursing infants, even with prolonged therapy, suggests that use of meropenem during breastfeeding is probably compatible. However, additional data are warranted and, until such data are available, infants should be monitored for the most common (2%) adverse effects observed in adult patients (headache, nausea, constipation, diarrhea, anemia, vomiting, and rash (1)). As cited in Meropenem-National Library of Medicine LactMed database, November 6, 2013. No teratogenic effects due to mesalamine have been described and, although toxicity in the fetus has been reported in one case, a causal relationship between the drug and the outcome is controversial. It also results from metabolism in the large intestine of the oral preparation sulfasalazine, which is split to mesalamine and sulfapyridine, and from balsalazide and olsalazine, oral formulations that are metabolized in the colon to mesalamine. The history, pharmacology, and pharmacokinetics of mesalamine and olsalazine were extensively reviewed in a 1992 reference (1). Reproduction studies in rats and rabbits at oral doses of 480 mg/kg/day observed no fetal toxicity or teratogenicity (2). Sulfasalazine and one of the metabolites, sulfapyridine, readily cross the placenta and could displace bilirubin from albumin if the concentrations were great enough. Mesalamine is bound to different sites on albumin than bilirubin and, thus, has no bilirubin-displacing ability (3). Moreover, only small amounts of mesalamine are absorbed from the cecum and colon into the systemic circulation, and most of this is rapidly excreted in the urine (4). The drug and metabolite levels (all in mcg/mL) and the number of patients were as follows: amniotic fluid (N = 4), 0. At delivery in a woman taking 1 g of mesalamine 3 times daily, the concentrations of the drug and its metabolite, 3. A review of drug therapy for ulcerative colitis recommended that women taking mesalamine to maintain remission of the disease should continue the drug when trying to conceive or when pregnant (7). Full-term deliveries occurred in 18, and 1 patient, with a history of 4 previous miscarriages, suffered a spontaneous abortion. Only one report has described possible in utero mesalamine-induced toxicity that may have occurred during 2nd trimester exposure to the drug (9). The term male infant had a serum creatinine at birth of 115 µmol/L (normal 1835 µmol/L). At this age, the serum creatinine was 62 µmol/L with a creatinine clearance of 52 mL/min (normal 8090 mL/min). A renal biopsy at 6 months of age showed focal tubulointerstitial lesions with interstitial fibrosis and tubular atrophy in the absence of cell infiltration (9). Because no other cause of the renal lesions could be found and there was some resemblance to lesions induced by the prostaglandin synthesis inhibitor, indomethacin, the authors attributed the defect to mesalamine (9). A letter published in response to this study, however, questioned the association between the drug and observed renal defect because of the lack of toxicity in an unpublished series of 60 exposed pregnancies and the lack of evidence that mesalamine causes renal prostaglandin synthesis inhibition in utero (10). A 1997 report described the successful outcomes of 19 pregnancies followed prospectively in 16 women with proven distal colitis (11). The women received either 4-g mesalamine enemas 3 times weekly or a 500-mg mesalamine suppository every night throughout gestation. No fetal abnormalities were observed during pregnancy and all of the full-term offspring were normal at birth and at a median follow-up of 2 years (range 2 months to 5 years) (11). There were no significant differences between the two groups in spontaneous abortions (6. There was one major defect (an extra right thumb) in the study group compared with five major anomalies in controls (ns). However, compared with controls, significantly more preterm deliveries occurred in subjects (13. Other agents used included 49 cases with prednisone, 101 with azathioprine or mercaptopurine, 27 with metronidazole, 18 with ciprofloxacin, and 2 with cyclosporine. The pregnancy outcomes of the first eligible pregnancy in the 113 women were 2 ectopic pregnancies, 2 elective abortions, 16 spontaneous abortions, 6 premature infants, 85 full-term infants (1 set of twins), and 3 major defects (2. However, one of the "defects" was a case of immature lungs, which is not considered a congenital defect.
Generic 125 mcg thyroxine with amex
Theoretically treatment diverticulitis discount thyroxine on line, exposure to agents in this class at the time of implantation could result in impaired fertility as a result of embryonic cytotoxicity, but this has not been studied in humans. No teratogenic effects were observed in rats and rabbits administered lamivudine up to approximately 130 and 60 times, respectively, the usual human adult dose. Early embryo lethality was observed in rabbits at doses close to those used in humans and above. Adverse effects on neurobehavior development in mice offspring resulting from a combination of lamivudine and zidovudine were described in a 2001 study (3). The effects on somatic and sensorimotor development were minor but more marked in exposed offspring than when either drug was given alone (2). Further, alterations of social behavior were observed in both sexes of exposed offspring (3). The low molecular weight (about 229) of lamivudine suggests that it will cross the placenta. A study published in 1997 described the human placental transfer of lamivudine using an ex vivo single cotyledon perfusion system (4). Confirming this, a 1998 study found that combination therapy with zidovudine did not affect the pharmacokinetics of lamivudine (5). Lamivudine freely crossed the placenta when given near term with nearly equivalent drug levels in the mother, cord blood, and newborn. The Antiretroviral Pregnancy Registry reported, for the period January 1989 through July 2009, prospective data (reported before the outcomes were known) involving 4702 live births that had been exposed during the 1st trimester to one or more antiretroviral agents (6). There were 8331 outcomes exposed to lamivudine (3314 in the 1st trimester and 5017 in the 2nd/3rd trimesters) in combination with other antiretroviral agents. There were 222 birth defects (96 in the 1st trimester and 126 in the 2nd/3rd trimesters). In reviewing the birth defects of prospective and retrospective (pregnancies reported after the outcomes were known) registered cases, the Registry concluded that, except for isolated cases of neural tube defects with efavirenz exposure in retrospective reports, there was no other pattern of anomalies (isolated or syndromic) (6). Lamivudine was taken by 29 women in various combinations that included zidovudine, nelfinavir, indinavir, stavudine, nevirapine, and saquinavir. The outcomes of the pregnancies included one stillbirth, one case of microcephaly, and five infants with birth weights <2500 g, two of whom were premature (7). Ten days later, she was started on a prophylactic regimen of lamivudine (300 mg/day), zidovudine (600 mg/day), and indinavir (2400 mg/day). The indinavir dose was reduced to 1800 mg/day, 4 weeks after the start of therapy because of the development of renal calculi. Several reports have described the apparent safe use of lamivudine, usually in combination with other agents, during human pregnancy (912). In one report, lamivudine concentrations in the newborn were similar to those in the mother (11). Mitochondrial disease is relatively rare in France (estimated prevalence 1 in 500020,000 children). From an ongoing epidemiological survey of 1754 motherchild pairs exposed to zidovudine and other agents during pregnancy, however, 8 children with possible mitochondrial dysfunction were identified. Four of the cases were exposed to zidovudine alone and four to a combination of zidovudine and lamivudine. Moreover, the toxicity may have been potentiated by combination of these agents (13). They also postulated this mechanism was involved in the development of a lipodystrophy syndrome of peripheral fat wasting and central adiposity, a condition that has been thought to be related to protease inhibitors (14). Although three had neurological symptoms, none had raised levels of lactate in the cerebrospinal fluid. Moreover, histological or histochemical features of mitochondrial disease were found in only two cases. He received postnatal prophylaxis with lamivudine and zidovudine for 4 weeks until the agents were discontinued because of anemia. Other adverse effects that were observed in the infant (age at onset) were hypocalcemia (shortly after birth), Group B streptococcal sepsis, ventricular extrasystoles, prolonged metabolic acidosis, and lactic acidemia (8 weeks), a mild elevation of long chain fatty acids (9 weeks), and neutropenia (3 months). The metabolic acidosis required treatment until 7 months of age, whereas the elevated plasma lactate resolved over 4 weeks. The elevated plasma fatty acid level was confirmed in cultured fibroblasts, but other peroxisomal functions (plasmalogen biosynthesis and catalase staining) were normal. The child was reported to be healthy and developing normally at 26 months of age (15). A case of life-threatening anemia following in utero exposure to antiretroviral agents was described in 1998 (16). Because of an inadequate response, didanosine was discontinued and lamivudine and zalcitabine were started in the 3rd trimester. At term, a pale, male infant was delivered who developed respiratory distress shortly after birth. Because no other cause of the anemia could be found, the authors attributed the condition to bone morrow suppression, most likely to zidovudine. A contribution of the other agents to the condition, however, could not be excluded (16). Two reviews, one in 1996 and the other in 1997, concluded that all women currently receiving antiretroviral therapy should continue to receive therapy during pregnancy and that treatment of the mother with monotherapy should be considered inadequate therapy (17,18). The same conclusion was reached in a 2003 review with the added admonishment that therapy must be continuous to prevent emergence of resistant viral strains (19). A review published in 2000 described seven clinical trials that have been effective in reducing perinatal transmission, five with zidovudine alone, one with zidovudine plus lamivudine, and one with nevirapine (22). Prolonged use of zidovudine in the mother and infant was the most effective for preventing vertical transmission, but also was the most expensive. The combination of lamivudine and zidovudine, consisting of antepartum, intrapartum, and postpartum maternal therapy with continued therapy in the infant for 1 week, may have been as effective as prolonged zidovudine (22). The following specific drugs have large enough groups of exposed women to warrant a separate analysis: abacavir, didanosine, efavirenz, lamivudine, lopinavir, nelfinavir, nevirapine, ritonavir, stavudine, tenofovir, and zidovudine. No increases in risk of overall birth defects or specific defects have been detected to date. For lamivudine and zidovudine sufficient numbers of 1st trimester exposures have been monitored to detect at least a 1. For abacavir, efavirenz, nelfinavir, nevirapine, ritonavir, and stavudine sufficient numbers of 1st trimester exposures have been monitored to detect at least a twofold increase in risk of overall birth defects. Although no pattern of birth defects has been detected with didanosine, the Committee continues to monitor the apparent increased frequency of defects among infants exposed to didanosine in the 1st trimester of gestation. To date, the Registry has not demonstrated an increased prevalence of birth defects overall, or in the specific classes studied, or among women exposed to abacavir, efavirenz, lamivudine, nelfinavir, nevirapine, ritonavir, stavudine, or zidovudine individually or in combination during the 1st trimester when compared with observed rates for "early diagnoses" in population-based birth defects surveillance systems. While the Registry to date has not detected a major teratogenic signal overall or within classes of drugs or the individual drugs analyzed separately, the population exposed and monitored to date is not sufficient to detect an increase in the risk of relatively rare defects. In 10 women on lamivudine monotherapy (300 mg/day), the mean drug concentrations in maternal serum and breast milk were 0. Pharmacokinetics and antiretroviral activity of lamivudine alone or when coadministered with zidovudine in human immunodeficiency virus type 1-infected pregnant women and their offspring. Combination antiretroviral therapy in human immunodeficiency virus-infected pregnant women. The use of human immunodeficiency virus postexposure prophylaxis after successful artificial insemination. Mitochondrial toxicity induced by nucleoside-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Mitochondrial and peroxisomal dysfunction following perinatal exposure to antiretroviral drugs. Reproduction studies with lamotrigine in two animal species showed evidence, in the presence of maternal toxicity, of developmental toxicity (growth restriction, behavioral deficits, and death) but not structural anomalies. Moreover, at least two reviews have concluded that this anticonvulsant may be associated with a lower risk of teratogenicity (1,2). However, a recent report has shown a significant risk for oral clefts following 1st trimester exposure (3). A 2012 review concluded that the absolute risk of lamotrigine-induced cleft lip and palate or cleft palate alone was between 0. In addition, a significant increase in the risk for major defects has been reported when lamotrigine was combined with valproate (5). It is also indicated for the maintenance treatment of bipolar disorder to delay the time to occurrence of mood episodes. Protein binding is moderate (about 55%) and the agent is metabolized to inactive metabolites (6).
Vanilla Leaf (Deertongue). Thyroxine.
- How does Deertongue work?
- Malaria.
- Dosing considerations for Deertongue.
- Are there safety concerns?
- Are there any interactions with medications?
- What is Deertongue?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96252
Generic thyroxine 50mcg without a prescription
If a pregnant woman is exposed to this drug treatment knee pain cheap thyroxine online master card, she should be informed of the absence of human pregnancy experience and the potential for embryofetal risk. It is indicated for adjunctive treatment of seizures associated with Lennox-Gastaut syndrome in children 4 years of age and adults. Only 34% of the drug is bound to plasma proteins, mostly to albumin, and the plasma half-life is about 610 hours (1). In rabbits, daily oral doses during organogenesis resulting in exposures that were >0. In prenatal and postnatal studies with rats, daily oral doses given from implantation through weaning resulting plasma exposures that were <0. Long-term exposures to rufinamide in mice and rats were carcinogenic, but the drug was not mutagenic or clastogenic in multiple assays. The low molecular weight (about 238), lipophilic nature, and long plasma half-life suggest that exposure of the embryofetus will occur. The low molecular weight (about 238), lipophilic nature, and long plasma half-life suggest that the drug will be excreted into breast milk, but the extensive metabolism should limit the amount. If mother chooses to nurse her infant while taking rufinamide, the infant should be closely observed for changes in behavior and other signs of toxicity. Somnolence, vomiting, and headache were the three most frequent adverse reactions observed in pediatric patients treated with the drug (1). The animal data suggested risk (reduced fetal weights and late resorptions in two species) but these effects occurred with doses that were maternally toxic. However, lower nonmaternal toxic doses in rats were associated with postimplantation losses. If the drug is indicated in a pregnant woman, she should be informed of the potential risk to her embryofetus. It is indicated for the treatment of patients with intermediate- or high-risk myelofibrosis, including primary myelofibrosis, postpolycythemia vera myelofibrosis, and postessential thrombocythemia myelofibrosis. The mean elimination half-life for ruxolitinib alone is about 3 hours, whereas it is about 5. At these doses, reduced fetal weights were noted in rats and rabbits, and increased late resorptions in rabbits, but maternal toxicity occurred in both species. Ruxolitinib was not carcinogenic in long-term studies in mice and rats, and was not mutagenic or clastogenic in multiple assays. It is not known if ruxolitinib or its two active metabolites cross the human placenta. The molecular weight of the parent drug (about 404) suggests that the drug will cross, but the high plasma protein binding and relatively short mean elimination half-lives should limit the exposure. The molecular weight of the parent drug (about 404) suggests that the drug, and possibly its two active metabolites, will be excreted into breast milk, but the high (97%) plasma protein binding and relatively short mean elimination half-lives (3 hours for the parent drug and 5. However, thrombocytopenia and anemia occurred in >20% of patients treated with the drug and >10% experienced bruising, dizziness, and headache (1). Thus, if the drug is given during breastfeeding, a nursing infant should be monitored for these adverse effects. The Calorie Control Council believes that the agent can be safely used by pregnant women (1). However, others recommended avoidance of saccharin or, at least, cautious use of it in pregnancy (24). Saccharin, a derivative of naphthalene, is absorbed slowly after oral ingestion and is rapidly and completely excreted, as the unmetabolized compound, by the kidneys. Although a large amount of medical research has been generated concerning saccharin, very little of this information pertains to its use by pregnant women or to its effect on the fetus (2,3). Fetal levels were still present 5 hours after the end of the infusion and 2 hours after maternal concentrations were undetectable. A study, published in 1986, documented that saccharin also crosses the placenta to the human fetus (6). Six diabetic women, consuming 25100 mg/day of saccharin by history, were delivered at 3642 weeks. No increase in the incidence of spontaneous abortions among women consuming saccharin has been found (9). In some animal species, particularly after second-generation studies, an increased incidence of bladder tumors was observed (3). However, epidemiologic studies have failed to associate the human use of saccharin with bladder cancer (3). Similarly, no evidence was found in a study of the Danish population that in utero saccharin exposure was associated with an increased risk of bladder cancer during the first 3035 years of life (3,10). However, at least one investigator believes that these studies must be extended much further before they are meaningful, because bladder cancer is usually diagnosed in the elderly (4). In six healthy women, saccharin, 126 mg/12 fluid ounces, contained in two commercially available soft drinks, was given every 6 hours for nine doses. After single or multiple doses, median peak concentrations of saccharin occurred at 0. Milk concentrations ranged from <2001056 ng/mL after one dose to 1765 ng/mL after nine doses. The amounts of saccharin a nursing infant could consume from milk were predicted to be much less than the usual intakes of children <2 years old (11). Prenatal development in the rat following administration of cyclamate, saccharin and sucrose. Abortifacient and emmenagogue effects have been suggested (1,2), but there apparently is no clinical evidence to support these effects when safflower oil is used as a food. The oil contains a high concentration of n-6 polyunsaturated fatty acids, including the essential linoleic acid. In addition, the seeds contain tracheloside (a lignan glycoside) and serotonin derivatives. In addition, capsules containing safflower oil have sometimes served as controls in clinical trials. The flowers have been used as yellow and red dyes since ancient times, as well as a flavoring agent in foods (1,2). Although mature human milk contains n-3 fatty acids (4), the probable small amounts of n-6 fatty acids excreted into milk when safflower oil is used as a food should not adversely affect a nursing infant. Although human data have been published and no congenital malformations attributable to salmeterol were observed, the data are too limited to assess the safety of salmeterol. Moreover, the above study lacked the sensitivity to identify minor anomalies because of the absence of standardized examinations. Late-appearing major defects may also have been missed because of the timing of the questionnaires. Moreover, salmeterol is more effective than doubling the dose of inhaled corticosteroids and may have advantages over theophylline in terms of effectiveness and tolerability (2). In addition, the very low or undetectable maternal plasma levels that occur with therapeutic inhaled doses suggest that the risk to the fetus from the drug is probably minimal or nonexistent. Because the drug acts locally in the lung, plasma levels are very low or undetectable and are a result of swallowed salmeterol. The fetal toxicity noted in rabbits was not thought to be relevant to humans because -agonists characteristically induce these effects in animals (4). In a study with pregnant rats, salmeterol concentrations in mammary tissue, placenta, and fetus after oral administration were comparable to those in maternal blood up to 6 hours after a dose (5). At 24 hours, the disposition of the drug in the fetus was primarily in the gastrointestinal tract. Although the molecular weight (about 604 for salmeterol xinafoate) is low enough, plasma levels after an inhaled therapeutic dose are very low or undetectable. A 1998 noninterventional observational cohort study described the outcomes of pregnancies in women who had been prescribed 1 of 34 newly marketed drugs by general practitioners in England (6). The outcomes of these pregnancies included 7 spontaneous abortions, 2 ectopic pregnancies, 4 elective abortions, 5 unknown outcomes, and 47 live births (3 premature). The molecular weight (about 604 for salmeterol xinafoate) is low enough for excretion into breast milk, but maternal plasma levels after an inhaled therapeutic dose are very low or undetectable. The herb is used by some cultures in Mexico and exposure during gestation may have occurred. However, because smoking the herb has caused toxic, persistent psychosis in an 18-year-old female and a 21-year-old male (13), it is best avoided in pregnancy. The hallucinogenic substances from the leaves and juices of the herb have been used by some groups in Mexico for healing and divinatory rituals. The hallucinogenic effects are produced by smoking or chewing the leaves or drinking the juices.
Buy thyroxine 50 mcg overnight delivery
However treatment action group discount 150mcg thyroxine with visa, three fetuses (one set of twins) exposed to naproxen at 30 weeks for 26 days in an unsuccessful attempt to halt premature labor had markedly decreased plasma concentrations of prostaglandin E. Use in other patients for premature labor at 34 weeks or earlier has not resulted in neonatal problems (23,24). Conservative management was initiated and the infant was improving by the sixth postnatal day. The authors acknowledged that further studies were needed because most of these associations had not been reported from other databases (26). Following 250 or 375 mg twice daily, maximum milk levels were found 4 hours after a dose and ranged from 0. The American Academy of Pediatrics classifies naproxen as compatible with breastfeeding (29). Prescription, over-the-counter, and herbal medicine use in a rural, obstetric population. Maternal drug use and infant cleft lip/palate with special reference to corticoids. Risk of congenital anomalies in pregnant users of nonsteroidal anti-inflammatory drugs: a nested case-control study. The effects of nonsteroidal antiinflammatory compounds on fetal circulation and pulmonary function. Persistent pulmonary hypertension and abnormal prostaglandin E levels in preterm infants after maternal treatment with naproxen. Analysis of nonsteroidal antiinflammatory drugs in meconium and its relation to persistent pulmonary hypertension of the newborn. Persistent pulmonary hypertension after maternal naproxen ingestion in a term newborn: a case report. Human pregnancy experience, however, is too limited to assess the embryofetal risk. The safety and effectiveness of naratriptan have not been established for cluster headaches. Plasma protein binding is low (28%31%) and the elimination halflife is 6 hours (2). Developmental toxicity was observed in pregnant rats and rabbits at oral doses producing maternal plasma drug levels as low as 11 and 2. Toxicity consisted of embryo lethality, minor fetal structural variations, pup mortality, and offspring growth restriction. The highest dose was associated with maternal toxicity as evidenced by decreased body weight gain. When naratriptan was administered before and throughout the mating period at 60 mg/kg/day, there was an increase in preimplantation loss. Impairment of fertility in both male (testicular effects) and female (anestrus) rats occurred at higher doses. Exposure to the two highest doses-60 and 340 mg/kg/day-late in gestation and during lactation resulted in offspring behavioral impairment (tremors) and decreased viability and growth (2). Pregnant rabbits received daily oral doses during organogenesis, which produced maternal plasma levels ranging from 2. Developmental toxicity, observed at all doses, consisted of embryonic death and fetal variations (major blood vessel variations, supernumerary ribs, and incomplete skeletal ossification). Fetal skeletal malformation (fused sternebrae) was observed in one rabbit subspecies at the highest dose. The molecular weight (about 336 for the free base), low plasma protein binding, and long elimination half-life suggest that the drug will cross. The Sumatriptan/Naratriptan/Treximet Pregnancy Registry, covering the period January 1, 1996, through April 30, 2009, has outcome data for 57 prospectively enrolled (reported before pregnancy outcome was known) pregnancies exposed to naratriptan. There were 52 with earliest exposure in the 1st trimester and 5 in the 2nd trimester. There were no birth defects in the five cases with earliest exposure in the 2nd trimester. Retrospective reports (reported after the outcome was known) are often biased (only adverse outcomes are reported). There were three retrospective reports of birth defects exposed in the 1st trimester. Review of all birth defects from prospective and retrospective reports revealed no signal or consistent pattern to suggest a common cause (4). The molecular weight (about 336 for the free base), low plasma protein binding (28%31%), and the long elimination half-life (6 hours) suggest that the drug will be excreted into breast milk. However, a brief 2010 review concluded that the minimal amounts of other triptans excreted into breast milk are insufficient to cause adverse effects in breastfeeding infants (6). Triptan exposure during pregnancy and the risk of major congenital malformations and adverse pregnancy outcomes: results from the Norwegian Mother and Child Cohort Study. However, a study described below did find that exposure in the second half of pregnancy impaired the immune system of two newborns, a finding similar to the effect observed in adults treated with the drug. In 2011, the World Congress of Gastroenterology stated, "The safety of natalizumab during pregnancy is unknown. Although human data show no increased risk of birth defects, the agent is too new for adequate supportive data and there are no drugs of similar mechanism with which to compare. Nevertheless, if the maternal condition indicates that the drug is required, it should not be withheld because of pregnancy. If natalizumab is used in pregnancy, physicians are encouraged to enroll patients in the Tysabri pregnancy exposure registry by calling 800-456-2255. Reproduction studies have been conducted in pregnant guinea pigs and cynomolgus monkeys. At this dose in both species, serum levels in fetal animals at delivery were approximately 35% of maternal levels. Offspring exposed in utero had no drug-related changes in the lymphoid organs and had normal immune response (2). No carcinogenic, clastogenic, or mutagenic effects were observed in various assays and tests conducted with natalizumab. A series of four research reports, conducted by the manufacturer, on the effects of natalizumab in guinea pigs and cynomolgus monkeys was published in 2009 (36). One concern was that the drug would interfere with pregnancy because -4 integrins and their ligands appear to be critically involved in mammalian fertilization, implantation, and placental and cardiac development (36). The results and conclusions of the four studies were similar to those described above. Natalizumab crosses the placentas of guinea pigs and monkeys producing fetal drug concentrations approximately 35% of maternal concentrations at birth (2). Because of the similarity to the human placenta and the very long elimination half-life, passage to the fetus should be expected, regardless of the very high molecular weight (about 149,000). Moreover, immune globulin crosses the human placenta in significant amounts near term (see Immune Globulin Intravenous). There was no increase in the rate of birth defects compared with the expected background occurrence. At 37 3/7 weeks, she gave birth to a healthy 3160-g baby girl with Apgar scores of 10 and 10. A preliminary report from the Tysabri (Natalizumab) Pregnancy Register was described in two references (10,11). As of May 2011, 341 pregnant patients had been prospectively enrolled with 277 known outcomes. Several other pregnancy outcomes following use of natalizumab were also described in one of the references (11). No structural anomalies were evident, but a minor intracerebral hemorrhage was detected by ultrasound that later resolved. At age of 6 weeks, the infant was admitted to the hospital with respiratory syncitial virus bronchiolitis that resolved within a few days with symptomatic treatment. Because adults treated with natalizumab have a significant decrease in T-lymphocyte chemotaxis, both infants were examined for this decrease. The impaired effects on the immune system were found in both infants at 2 weeks of age but, in both cases, the impairment had resolved by 12 weeks of age. The authors concluded that the clinical relevance of the impaired chemotaxis in the context of host defense, especially against viral pathogens, was unclear because chemotaxis was just one of several features of altered immune cell functions under treatment with natalizumab (12). The molecular weight is very high (about 149,000), but immunoglobulins are excreted into breast milk.

Discount 50mcg thyroxine free shipping
A possible association between phenytoin and the rare defect symptoms 10 dpo order thyroxine amex, holoprosencephaly, was reported in 1993 (56). Because of psychomotor and petit mal seizures, the mother was treated with phenytoin (350 mg/day) and primidone (500 mg/day) during gestation. The female infant was born after 36 weeks of gestation with both weight and length above the 50th percentile. Malformations evident on examination were microcephaly, narrow forehead, hypertelorism, hypoplastic midface with anteverted nostrils, smooth philtrum, and thin vermillion border lip. Ultrasound examination revealed a right-sided renal duplication, hepatomegaly, a mild ventricular septal defect, and bilateral hip dysplasia. A partial lobar holoprosencephaly with ventral fusion of the cerebral hemispheres was noted on magnetic resonance imaging of the brain. Coarse gyri and horizontal cleavage, but no sagittal cleavage, was noted in the frontal lobe. Authors of correspondence relating to the above report noted that they had also identified a case of holoprosencephaly in a stillborn infant exposed in utero to anticonvulsants (59). Based on their experience, however, establishing an association between phenytoin and the defect would be very difficult because of the infrequent in utero exposure to anticonvulsants (1:250 births), the even lower frequency of exposure to phenytoin monotherapy (1:844 births), and the rarity of holoprosencephaly (1:10,000) (59). Thanatophoric dwarfism was found in a stillborn infant exposed throughout gestation to phenytoin (200 mg/day), phenobarbital (300 mg/day), and amitriptyline (>150 mg/day) (60). The cause of the malformation could not be determined, but drug and genetic causes were considered. Among these, exposure to monotherapy occurred in the following: phenytoin (N = 24), phenobarbital (N = 65), mephobarbital (N = 10), carbamazepine (N = 46), valproic acid (N = 80), and other agents (N = 16). No statistically significant associations were found with phenytoin monotherapy or polytherapy. Although the study confirmed some previously known associations, several new associations with anticonvulsants were discovered that require independent confirmation (see also Carbamazepine, Mephobarbital, Phenobarbital, and Valproic Acid) (61). Among 68 prospectively enrolled pregnancies exposed to phenytoin and lamotrigine, with or without other anticonvulsants, 62 were exposed in the 1st trimester resulting in 56 live births, 1 birth defect, 1 spontaneous abortion, and 4 elective abortions. There were six exposures in the 2nd/3rd trimesters resulting in six live births without birth defects (62). Twelve case reports have been located that, taken in sum, suggest phenytoin is a human transplacental carcinogen (1828,63). A 1989 study, however, found no in utero exposures to phenytoin among 188 cases of childhood neuroblastoma diagnosed between 1969 and 1988 at their center (64). Hemorrhage occurs during the first 24 hours after birth and may be severe or even fatal. The exact mechanism of the defect is unknown but may involve phenytoin induction of fetal liver microsomal enzymes that deplete the already low reserves of fetal vitamin K (79). Phenytoin-induced thrombocytopenia has also been reported as a mechanism for hemorrhage in the newborn (76). They proposed early vitamin K supplementation of at-risk pregnancies to prevent this disfiguring malformation. Liver damage was observed in an infant exposed during gestation to phenytoin and valproic acid (81). Although they were unable to demonstrate which anticonvulsant caused the injury, the authors concluded that valproic acid was the more likely offending agent. Phenytoin may induce folic acid deficiency in the epileptic patient by impairing gastrointestinal absorption or by increasing hepatic metabolism of the vitamin (8284). Whether phenytoin also induces folic acid deficiency in the fetus is less certain because the fetus seems to be efficient in drawing on available maternal stores of folic acid (see Folic Acid). Low maternal folate levels, however, have been proposed as one possible mechanism for the increased incidence of defects observed in infants exposed in utero to phenytoin. In a 1984 report, two investigators studied the relationship between folic acid, anticonvulsants, and fetal defects (82). In the retrospective part of this study, a group of 24 women treated with phenytoin and other anticonvulsants produced 66 infants, 10 (15%) with major anomalies. Two of the mothers with affected infants had markedly low red blood cell folate concentrations. A second group of 22 epileptic women was then supplemented with daily folic acid, 2. This group produced 33 newborns (32 pregnancies-1 set of twins) with no defects, a significant difference from the nonsupplemented group. Loss of seizure control caused by folic acid lowering of phenytoin serum levels, which is known to occur, was not a problem in this small series (82). Negative associations between phenytoin and folate deficiency have been reported (83,84). In one study, mothers were given supplements with an average folic acid dose of 0. Defects were observed in 20 infants (15%) from the 133 women taking anticonvulsants, which is similar to the reported frequency in pregnant patients not given supplements. The effects of exposure (at any time during the 2nd or 3rd month after the last menstrual period) to folic acid antagonists on embryofetal development were evaluated in a large, multicenter, casecontrol surveillance study published in 2000 (85). The case subjects were 3870 infants with cardiovascular defects, 1962 with oral clefts, and 1100 with urinary tract malformations. The risk of malformations in control infants would not have been reduced by vitamin supplementation, and none of the controls used folic acid antagonists. The pharmacokinetics and placental transport of phenytoin have been extensively studied and reviewed (8688). Animal studies and recent human reports suggest a dose-related teratogenic effect of phenytoin (89,90). Although these results are based on a small series of patients, it is reasonable to avoid excessively high plasma concentrations of phenytoin. Close monitoring of plasma phenytoin concentrations is recommended to maintain adequate seizure control and prevent potential fetal hypoxia. No effect was detected from phenytoin as measured by serum human placental lactogen, 24-hour urinary total estriol excretion, placental weight, and birth weight. In a study evaluating thyroid function, no differences were found between treated epileptic pregnant women and normal pregnant controls (92). Thyroxine levels in the cord blood of anticonvulsant-exposed infants were significantly lower than in controls, but this was shown to be caused by altered protein binding and not altered thyroid function. Other parameters studied-thyrotropin, free thyroxine, and triiodothyronine-were similar in both groups. The effect of phenytoin on maternal and fetal vitamin D metabolism was examined in a 1984 study (93). In comparison to normal controls, several significant differences were found in the level of various vitamin D compounds and in serum calcium, but the values were still within normal limits. The authors doubted whether the observed differences were of major clinical significance. Phenytoin may be used for the management of digitalis-induced arrhythmias that are unresponsive to other agents and for refractory ventricular tachyarrhythmias (9496). This short-term use has not been reported to cause problems in the exposed fetuses. The drug has also been used for anticonvulsant prophylaxis in severe preeclampsia (97). In a 1988 study designed to evaluate the effect of in utero exposure to anticonvulsants on intelligence, 148 Finnish children of epileptic mothers were compared with 105 controls (99). In those mothers treated during pregnancy, 103 received phenytoin (monotherapy in 54 cases), all during the first 20 weeks. Two of the 148 children of epileptic mothers were diagnosed as mentally deficient and 2 others had borderline intelligence (the mother of 1 of these latter children had not been treated with anticonvulsant medication). However, the presence of hypertelorism and digital hypoplasia, two minor anomalies considered typical of exposure to phenytoin, was not predictive of low intelligence (99). The verbal comprehension and expressive language scores were also significantly lower (0. Major malformations were observed in two phenytoinexposed children (cleft palate and hypospadias; meningomyelocele and hydrocephalus), none of the phenytoin controls, two carbamazepine-exposed children (missing last joint of right index finger and nail hypoplasia; hypospadias), and one carbamazepine control (pulmonary atresia). The study results suggested that phenytoin had a clinically important negative effect on neurobehavioral development that was independent of maternal or environmental factors (100).
