

6.25mg carvedilol with amex
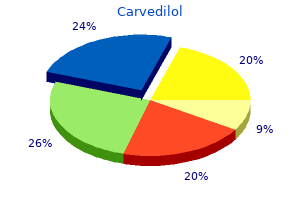
The results strongly support earlier work indicating that terrestrial vertebrates are more closely related to the lungfish than to the coelacanth blood pressure medication extreme tiredness purchase genuine carvedilol line. Molecular Clocks Measure the Rate of Evolutionary Change In many cases, we would like to estimate not only which members of a set of species are most closely related, but also when their common ancestors lived. The ability to construct phylogenetic trees from protein and nucleic acid sequences led to the development of molecular clocks, which use the rate of change in amino acid or nucleotide sequences as a way to estimate the time of divergence from a common ancestor. Phylogenetic evidence indicates that the lungfish (a) and not the coelacanth (b) is a common ancestor of amphibians, reptiles, birds, and mammals. In both cases, changes in amino acid sequence and nucleotide sequence increase linearly with time. The results show that humans and zebrafish last shared a common ancestor about 450 million years ago, and humans last shared a common ancestor with chimpanzees about 7-10 million years ago. The Complex Origins of the Human Genome Current fossil, molecular, and genomic evidence indicates that our species, Homo sapiens, arose in Africa about 300,000 years ago from earlier species of Homo. The genomes of two extinct groups who lived in the Middle East, Asia, and Europe, the Neanderthals and the Denisovans, have been sequenced and compared with the genomes of present-day humans. The results show that modern human populations outside Africa, including those of the Middle East, Europe, Asia, Australia/Oceania, and the Americas, carry sequences from these two groups. We know quite a lot about the genome of the Neanderthals and the contributions they made to our genome. The first Neanderthal genome was assembled in 2010 from three skeletons discovered in a Croatian cave. Comparative genome analysis shows that the genomes of our species and the Neanderthals are the same size (about 3. For at least 30,000 years, Neanderthals coexisted with anatomically modern humans (H. In fact, gene flow from extinct Neanderthals to modern humans through interbreeding is estimated to represent about 2 percent of the genome of non-African populations. However, different individuals carry different portions of the Neanderthal genome; taken together, upward of 20 percent of the Neanderthal genome may be present in the genomes of modern non-African populations. Second, Neanderthals and members of our species did interbreed, and Neanderthals contributed to our genome. In (a) the data are calculated by divergence times based on the fossil record, and in (b) the data are based on synonymous nucleotide substitutions, which are mutations that do not result in any changes in the amino acid sequence of a protein. A nuclear Denisovan genome sequence shows that they are more closely related to Neanderthals than to our species, and that Denisovans and Neanderthals separated from a common ancestral species more than 430,000 years ago. In addition, the Denisovan genome contains sequences from another, as yet unknown, archaic group that made no contribution to the Neanderthal genome. The Neanderthal and Denisovan genomes were assembled from fossil remains that are 40,000 to 80,000 years old. The recent sequencing of a genome from a 700,000-year-old horse fossil opens the possibility that genome sequences can be recovered from fossils of much older human species and used to identify the archaic species that contributed to the Denisovan genome. For now, using the paleogenomic techniques currently available, we can expect exciting answers to questions about the similarities and differences between our genome and those of other human species, providing revolutionary insights into the evolution of our species and other human species that preceded us on this planet. The latter two groups branched off from our last common ancestor before our species left Africa. Genomic analysis shows that there was interbreeding between members of our species with Neanderthals and Denisovans, making our genome a mosaic with contributions from at least two other human species. More genetic identity exists between two persons chosen at random from a human population than between two chimpanzees chosen at random from a chimpanzee population. Interestingly, about 90 percent of the genetic differences that do exist occur between individuals rather than between populations. This unusually high degree of genetic relatedness in all humans supports the idea that our species arose recently from a small founding group of individuals. Other genetic data show that the highest levels of human genetic variation occur within African populations. As with any explanation of human origins, the out-of-Africa hypothesis is actively debated. Some data suggest two or more out-of-Africa dispersals, as well as different timings of dispersals and migration routes. Your Turn pproximately 2 million years ago, a large-brained, tool-using hominid called Homo erectus appeared in East Africa. These groups disappeared 50,000 to 30,000 years ago-around the same time that anatomically modern humans (H. At present, the most widely accepted hypothesis explaining the presence of anatomically modern humans is the outof-Africa hypothesis. This hypothesis is based on genetic data derived from mitochondrial, Y chromosome, and whole-genome sequencing of both archaic hominin fossils and modern human populations. Some genetic and archaeological evidence appears to support two separate dispersals of humans out of Africa. What are these data, and how might they be reconciled with the single-dispersal hypothesis Given that genetic studies show that all people on Earth are remarkably similar genetically, how did we come to develop the concept of racial differences How has modern genomics contributed to the debate about the validity and definition of the term "race" For an interesting discussion of race, human variation, and genomics, see Lewontin, R. Confusion about human races, on the Social Sciences Research Center Web site (raceandgenomics. Tyrosinemia is caused by the lack of an enzyme in the degradation pathway of the amino acid tyrosine. Accumulation of metabolic intermediates causes progressive liver dysfunction and kidney problems. She faces a lifetime of antirejection drug therapy and may require a kidney transplant. In the United States, tyrosinemia occurs in only 1/100,000 births, and in this case, two states made different decisions about newborn testing for this disorder. In a region of Quebec, Canada, 1 in 22 people are heterozygous for the mutant tyrosinemia allele. Using the frequency of heterozygotes, calculate the frequency of recessive homozygotes in this population. Critics argue that a uniform panel of disorders should be used by all states in newborn testing. Others argue that the current testing system should be replaced by whole genome sequencing for all newborns. In some cases, this can be observed at the phenotypic level, but analysis at the amino acid level and especially the nucleotide level provides a more direct way to estimate genetic variation. In addition, this method can be used to calculate the frequency of heterozygotes for a given gene in a population. Natural selection changes allele frequency in populations leading to evolutionary change. Selection for quantitative traits can involve directional selection, stabilizing selection, or disruptive selection. In addition to natural selection, other forces act on allele frequencies in populations. These include mutation, migration, and Mastering Genetics For activities, animations, and review quizzes, go to the Study Area. Nonrandom mating alters genotype frequencies but does not change allele frequencies. The formation of new species depends on the formation of subpopulations and the accumulation of enough genetic differences that, when reunited, members of the subpopulations cannot interbreed. Phylogenetic analysis using morphology, amino acid sequences, or nucleotide sequences can be used to construct phylogenetic trees showing the evolutionary relationships among a group of organisms. When calibrated with molecular clocks, the evolutionary changes on a phylogenetic tree can be calibrated with a time scale. Among Ashkenazi Jews of Central European ancestry, about 1 in 3600 children is born with the disease.
Generic 25mg carvedilol
The genes encoding the digestive enzymes trypsin and chymotrypsin are examples pulse pressure readings carvedilol 6.25mg with visa, as are those that encode the respiratory molecules myoglobin and the various forms of hemoglobin. We conclude that the genes arose from a common ancestral gene through duplication. During evolution, the related genes diverged sufficiently that their products became unique. Other support includes the presence of gene families- groups of contiguous genes whose products perform the same, or very similar functions. Humans have at least four similar copies of the gene, while all nonhuman primates have only a single copy. These evolutionary periods coincide with the emergence of the human lineage in primates. The function of these genes has now been related to the enhancement of dendritic spines in the brain, which is believed to contribute to the evolution of expanded brain function in humans, including the development of language and social cognition. Other examples of gene families arising from duplication during evolution include the various types of human hemoglobin polypeptide chains (Chapter 14), the immunologically important T-cell receptors and antigens encoded by the major histocompatibility complex, and the clusters of multiple Hox genes that are important during development in vertebrates (see Chapter 23). By forming a chromosomal loop prior focused on finding associations with human diseases. If the centromere ciated with autism and other neurological disorders, and is not part of the rearranged chromosome segment, it is with cancer. If the centromere is part of ated with Type I diabetes and cardiovascular disease. Relevant to this chapter, these findings reveal and one noninverted homolog are called inversion that duplications and deletions are no longer restricted heterozygotes. Two such chromosomes in meiosis can be to textbook examples of these chromosomal mutations. We will return to this interesting topic later in the text (see If crossing over does not occur within the inverted segChapter 21), when genomics is discussed in detail. However, if crossing over does occur within the inversion loop, abnormal chromatids are 8. In any meiotic tetrad, a single crossover between nonThe inversion, another class of structural variation, is sister chromatids produces two parental chromatids and a type of chromosomal aberration in which a segment two recombinant chromatids. When the crossover occurs of a chromosome is turned around 180 degrees within within a paracentric inversion, however, one recombinant a chromosome. An inversion does not involve a loss of dicentric chromatid (two centromeres) and one recomgenetic information but simply rearranges the linear gene binant acentric chromatid (lacking a centromere) are sequence. Both contain duplications and deletions of chrothe length of the chromosome and subsequent reinsertion mosome segments as well. This polarized movement produces dicentric bridges that are cytologically recognizable. A dicentric chromatid usually breaks at some point so that part of the chromatid goes into one gamete and part into another gamete during the reduction divisions. Therefore, gametes containing either recombinant chromatid are deficient in genetic material. When such a gamete participates in fertilization, the zygote most often develops abnormally, if at all. The recombinant chromatids that are directly involved in the exchange have duplications and deletions. In plants, gametes receiving such aberrant chromatids fail to develop normally, leading to aborted pollen or ovules. In animals, the gametes have developed prior to the meiotic error, so fertilization is more likely to occur in spite of the chromosome error. However, the end result is the production of inviable embryos following fertilization. Because offspring bearing crossover gametes are inviable and not recovered, it appears as if the inversion suppresses crossing over. Actually, in inversion heterozygotes, the inversion has the effect of suppressing the recovery of crossover products when chromosome exchange occurs within the inverted region. If crossing over always occurred within a paracentric or pericentric inversion, 50 percent of the gametes would be ineffective. Furthermore, up to one-half of the viable gametes have the inverted chromosome, and the inversion will be perpetuated within the species. Evolutionary Advantages of Inversions Because recovery of crossover products is suppressed in inversion heterozygotes, groups of specific alleles at adjacent loci within inversions may be preserved from generation to generation. If the alleles of the involved genes confer a survival advantage on organisms maintaining them, the inversion is beneficial to the evolutionary survival of the species. When an organism is heterozygous for a balancer chromosome, desired sequences of alleles are preserved during experimental work. The key to its solution is to draw out the tetrad and follow the chromatids undergoing a double crossover. Reciprocal translocation, for example, involves the exchange of segments between two nonhomologous chromosomes. The least complex way for this event to occur is for two nonhomologous chromosome arms to come close to each other so that an exchange is facilitated. If the exchange includes internal chromosome segments, four breaks are required, two on each chromosome. The genetic consequences of reciprocal translocations are, in several instances, similar to those of inversions. The presence of a translocation does not, therefore, directly alter the viability of individuals bearing it. Homologs that are heterozygous for a reciprocal translocation undergo unorthodox synapsis during meiosis. As with inversions, some of the gametes produced are genetically unbalanced as a result of an unusual alignment during meiosis. In the case of translocations, however, aberrant gametes are not necessarily the result of crossing over. The chromosome with centromere 2 moves to the other pole, along with the chromosome containing either centromere 3 or centromere 4. The 1,4 combination contains chromosomes that are not involved in the translocation and are normal. Note that a third type of arrangement, where homologous centromeres segregate to the same pole during meiosis (called adjacent segregation-2), has not been included in this figure. This type of segregation has an outcome similar to adjacent segregation-1, with meiotic products containing genetically unbalanced duplicated and deleted chromosomal material. When genetically unbalanced gametes participate in fertilization in animals, the resultant offspring do not usually survive. Fewer than 50 percent of the progeny of parents heterozygous for a reciprocal translocation survive. This condition in a parent is called semisterility, and its impact on the reproductive fitness of organisms plays a role in evolution. In humans, such an unbalanced condition results in partial monosomy or trisomy, leading to a variety of birth defects. Cytogenetic studies of the parents and their offspring from these unusual cases explain the cause of familial Down syndrome. That is, one parent has the majority of chromosome 21 translocated to one end of chromosome 14. This individual is phenotypically normal, even though he or she has only 45 chromosomes and is referred to as a balanced translocation carrier. During meiosis in such an individual, one of the gametes contains two copies of chromosome 21-a normal chromosome and a second copy translocated to chromosome 14. When such a gamete is fertilized by a standard haploid gamete, the resulting zygote has 46 chromosomes but three copies of chromosome 21. Other potential surviving offspring contain either the standard diploid genome (without a translocation) or the balanced translocation like the parent. Although not illustrated, adjacent-2 segregation is also thought to occur, but rarely.
Discount carvedilol 25mg without prescription
It is a natural process involved in morphogenesis and a protective mechanism against cancer formation blood pressure medication refills buy carvedilol australia. Individuals with two copies of a tumor suppressor gene would need to experience separate mutations in both copies to develop cancer, whereas individuals with only one functional copy (plus one mutant copy) would only need a single mutation. Therefore, it make sense that those who inherit one mutant copy of a recessive tumor-suppressor gene will have the higher risk of developing cancer. Mutations that produce oncogenes alter gene expression either directly or indirectly and act in a dominant capacity. Proto-oncogenes are those that normally function to promote or maintain cell division. In the mutant state (oncogenes), they induce or maintain uncontrolled cell division; that is, there is a gain of function. Generally, this gain of function takes the form of increased or abnormally continuous gene output. On the other hand, loss of function is generally attributed to mutations in tumor-suppressor genes, which function to halt passage through the cell cycle. To encourage infected cells to undergo growth and division, viruses often encode genes that stimulate growth and division. Many viruses either inactivate tumor-suppressor genes of the host or bring in genes that stimulate cell growth and division. By inactivating tumor-suppressor genes, the normal braking mechanism of the cell cycle is destroyed. Through mutation, such protective mechanisms are compromised in cancer cells, and as a result they show higher than normal rates of mutation, chromosomal abnormalities, and genomic instability. As such, they can activate or silence whole chromosomes (X chromosome, for example) or certain chromosomal regions; they can be responsible for parental imprinting; and they can influence gene activity in heterochromatin. Patterns of nucleotide demethylation and hypermethylation are often different when cancer cells are compared to normal cells, as are histone modifications. In addition, it is possible that genetic tests will not detect all breast cancer mutations. Proteases in general and serine proteases, specifically, are considered tumor-promoting agents because they degrade proteins, especially those in the extracellular matrix. Consistent with this observation are numerous observations that metastatic tumor cells are associated with higher than normal amounts of protease expression. As with many forms of cancer, a single gene alteration is not the only requirement. It is also possible that the transcripts produced do not result in a functional fusion protein. This could occur either by a nucleotide change from C to T on the coding strand or by a change from G to A on the template strand. Three circumstances described in the article include transfer of cancerous cells during an organ transplant, transfer of cancer from mother to fetus during pregnancy, and transfer of pathogens that are linked to cancer formation. Refer to the article for a more detailed description of these routes to cancer formation. One condition shared by these routes is the weakened immune system of the recipient. The frequency distribution in the backcross would be: 1/16 4/16 6/16 4/16 1/16 = = = = = 40 cm 37. We can say that each gene (additive allele) provides an equal unit amount to the phenotype, and the colors differ from each other in multiples of that unit amount. Even though correlation does not mean cause and effect, it would seem logical that as you increased fiber length, you would also increase fleece weight. It is probably safe to say that the increase in fleece weight is directly related to an increase in fiber length. A higher concordance value for monozygotic twins indicates a significant genetic component for a given trait. However, for measles and handedness, the difference is not as significant, indicating a greater role of the environment. Hair color has a significant genetic component, as do idiopathic epilepsy, schizophrenia, diabetes, allergies, cleft lip, and club foot. The genetic component of mammary cancer is present but minimal according to these data. Since there is a difference of 24 cm between the extremes, 24 cm/8 = 3 cm for each increment (each of the additive alleles). Your essay should include a description of various ratios typical of Mendelian genetics as compared with the more blending, continuously varying expressions of neoMendelian modes of inheritance. Either method indicates there are two alleles at each locus, for a total of four alleles. For the data in the table, it would appear that ridge count and height have the highest heritability values. Many traits, especially those we view as quantitative, are likely to be determined by a polygenic mode with possible environmental influences. The following are some common examples: height, general body structure, skin color, and perhaps most common behavioral traits, including intelligence. Another key factor in rapid response to selection often relates to the number of loci involved. If there are few loci, each with large phenotypic effects controlling a trait, response to selection is usually high. Finally, if flower size is not genetically correlated with other floral traits, size alone may not be subject to strong stabilizing selection, which would reduce genetic variation. Therefore, whereas other floral traits may show low response to selection, size alone may be more responsive. Genetic variability that provides rapid adjustments to changing environments is an evolutionary advantage. Therefore, in general, one would expect that high heritability would contribute to high evolutionary potential. Cosegregation is said to occur when the phenotypic trait and the molecular marker are genetically linked. The solution to these types of problems rests on determining the ratio of individuals expressing the extreme phenotype to the total number of individuals. If there are three gene pairs, the ratio is 1:64; four gene pairs, 1:256; or five gene pairs, 1:1024. Therefore, these data indicate that four gene pairs influence size in these guinea pigs. Note that there are additional possible parental genotypes that will yield F1 individuals that are heterozygous at all four loci. The frequency of aa types is determined by dividing the number of nontasters (37) by the total number of individuals (125). Know- B-39 ing the frequencies of all the genotypes, one can calculate p as the sum of 0. Since the frequencies of all the genotypes are known, one can calculate p as the sum of 0. Notice that there are more heterozygotes and fewer homozygotes (especially the ss types) than predicted in the population. For each of these values, one merely takes the square root to determine q, then computes p, and then "plugs" the values into the 2pq expression. Because a dominant lethal gene is highly selected against, it Solutions to Problems and Discussion Questions 2. Your essay should include a discussion of the original sources of variation coming from mutation and that migration can cause gene frequencies to change in a population if the immigrants have different gene frequencies compared to the host population. You should also describe selection as resulting from the biased passage of gametes and offspring to the next generation. The classification of organisms into different species is based on evidence (morphological, genetic, ecological, etc. That is, there must be evidence that gene flow does not occur among the groups being called different species. Indeed, classification above the species level is somewhat arbitrary and based on traditions that is unlikely that it will exist at too high a frequency, if at all.
Order genuine carvedilol online
Subsequent mutations allow cells of the tumor to become metastatic blood pressure medication video buy carvedilol 6.25 mg on line, spreading the cancer to other locations in the body where new malignant tumors appear. However, converging lines of evidence are now clarifying the importance of epigenetic changes in the initiation and maintenance of malignancy. These findings are helping researchers understand the properties of cancer cells that are difficult to explain by the action of mutant alleles alone. Evidence for the role of epigenetic changes in cancer has established epigenomic changes as a major pathway for the formation and spread of malignant cells. These studies showed that genomic hypomethylation is a property of all cancers examined to date. It also relaxes control over imprinted genes, causing cells to acquire new growth properties. Despite the fact that cancer cells are characterized by global hypomethylation, selected regions of their genome are hypermethylated when compared to normal cells. Selective hypermethylation of promoter-associated CpG islands silences certain genes, including tumor-suppressor genes, often in a tumor-specific fashion (Table 19. Because both alleles are inactivated (although by different mechanisms), cells are able to escape control of the cell cycle and divide continuously. Even more striking, in ovarian cancer, mutations in nine specific genes are predominant, but promoter hypermethylation is observed in 168 genes. These genes are epigenetically silenced, and their reduced expression is linked to the development and maintenance of this cancer. In sum, several lines of evidence support the role of epigenetic alterations in cancer: 1. Global hypomethylation may cause genomic instabil- Me Me Genetic mutation Me Me Me Me ity and the large-scale chromosomal changes that are a characteristic feature of cancer. Several mechanisms can cause the loss or silencing of the second allele: mutation, chromosomal aberration, or an epimutation. The second allele can be lost through genetic mutation, chromosomal aberration, or silencing by an epigenetic event. Epigenetic modifications can silence multiple genes, making them more effective in transforming normal cells into malignant cells than sequential mutations of single genes. In fact, many of the mechanisms that cause epigenetic changes in cancer cells are not well understood, partly because the changes take place very early in the conversion of a normal cell to a cancerous one, and partly because by the time the cancer is detected, alterations in methylation patterns have already occurred. Mutations in components of chromatin remodeling complexes and the histone modification system allow cells to escape cell-cycle control and divide continuously. In addition to abnormal regulation of methylation, many cancers also have altered patterns of chromatin remodeling. One form of remodeling is controlled by the reversible covalent modification of histone proteins in nucleosome cores. Abnormal regulation of each of these enzyme classes results in disrupted histone profiles and is associated with a variety of cancer subtypes. Histone acetylation is strongly correlated with activation of transcription (Chapter 17). Abnormalities in histone deacetylation have been identified as an early event in the transformation of normal cells into cancer cells. Because there are a large number of different proteins in this complex, specific subunit mutations are associated with specific cancers. Mutations within tumor cells can be homozygous or, in most cases, heterozygous, making them dosage sensitive. In summary, several lines of evidence support the role of epigenetic alterations in cancer: (1) epigenetic mechanisms can replace mutations as a way of silencing individual tumor-suppressor genes or activating oncogenes; (2) global hypomethylation may cause genomic instability and the large-scale chromosomal changes that are a characteristic feature of cancer; and (3) epigenetic modifications can silence multiple genes, making them more effective in transforming normal cells into malignant cells than sequential mutations of single genes. The approved epigenetic drugs are only moderately effective on their own and are best used in combination with other anticancer drugs. To develop more effective drugs, several important questions remain to be answered: What causes cancer cells to respond to certain epigenetic drugs Which combinations of chromatin remodeling drugs, histone modification drugs, and conventional anticancer drugs are most effective on specific cancers Which epigenetic markers will be effective in predicting sensitivity or resistance to newly developed drugs Further research into the mechanisms and locations of epigenetic genome modification in cancer cells will allow the design of more potent drugs to target epigenetic events as a form of cancer therapy. Epigenetic cancer therapy is focused on reprogramming gene expression through the use of drugs that alter events in chromatin remodeling in order to change the pattern of gene expression from malignant to normal. The focus of epigenetic therapy in the development of first-generation drugs has been the reactivation of genes silenced by methylation or histone modification, essentially reprogramming the pattern of gene expression in cancer cells. Food and Drug Administration, and another 18 or more drugs are in clinical trials. One approved drug, Vidaza (azacytidine), is used in the treatment of myelodysplastic syndrome, a precursor to leukemia, and acute myeloid leukemia. Environmental Induction of Epigenetic Change Environmental agents including nutrition, exposure to chemicals, and physical factors, such as temperature, can alter gene expression by affecting the epigenome. In the period immediately after the famine, mortality rates in this population doubled, and most of this increase was attributed to malnutrition. Studies were conducted for decades afterward on the health of adult children of women who were pregnant or became pregnant during the famine. Overall, the findings show that the severity of health effects was correlated with prenatal time of exposure to famine conditions. Adults who were exposed early in prenatal development (an F1 generation) had higher rates of several disorders-including obesity, heart disease, and breast cancer-and higher mortality rates than adults exposed later in development. In addition, as adults, there was increased risk for schizophrenia and other neuropsychiatric disorders for those with early exposure, perhaps related to nutritional deficiencies during development of the brain and nervous system. Some effects persisted in the F2 generation, where adults had abnormal patterns of growth and increased rates of obesity. Other studies in China and Africa on the adult children of women who were pregnant or became pregnant during times of famine confirm the deleterious impact of poor maternal nutrition during pregnancy on their offspring and subsequent generations. More direct evidence for the role of environmental factors in modifying the epigenome comes from studies in experimental animals. A low-protein diet fed to pregnant rats causes changes in the expression of several genes in both the F1 and F2 offspring. Increased expression of these genes is associated with hypomethylation of their promoters. Other evidence indicates that epigenetic changes triggered by this diet modification were gene specific. Another dramatic example of how epigenome modifications affect the phenotype comes from the study of coat color in mice, where color is controlled by the dominant allele Agouti (A). A nonlethal mutant allele (Avy) causes yellow pigment formation along the entire hair shaft, resulting in yellow fur color. This allele is the result of the insertion of a transposable element near the transcription start site of the Agouti gene. In these mice, coat colors range from yellow (unmethylated promoter) to pseudoagouti (highly methylated promoter). In addition to a gradation in coat color, there is also a gradation in body weight. Yellow mice are more obese than the brown pseudoagouti mice and are more likely to be diabetic. Alleles such as Avy that show variable expression from individual to individual in genetically identical strains caused by different patterns of epigenetic modifications to the alleles are called metastable epialleles. In other words, the epigenetic modifications to the Avy allele can be passed on to offspring; this is an example of transgenerational inheritance. To evaluate the role of environmental factors in modifying the epigenome, the diet of pregnant Avy mice was supplemented with methylation precursors, including folic acid, vitamin B12, and choline. In the offspring, variation in coat color was reduced and shifted toward the pseudoagouti (highly methylated) phenotype. In mice, two regions of the brain show preferential expression of maternal or paternal alleles. Upward of 1000 genes in the developing brain are imprinted, supporting the idea that epigenetic mechanisms operating in different regions of the brain may represent a major form of behavioral regulation. In humans, epigenetic changes have been documented during the progression of neurodegenerative disorders and in neuropsychiatric diseases, both of which show altered behavioral phenotypes. Epigenetic changes to the nervous system occur in Alzheimer disease, Parkinson disease, Huntington disease, and in schizophrenia and bipolar disorder. However, because the phenotypes in these disorders are influenced by a number of factors including genetic predispositions, events in prenatal development, and prenatal and postnatal environmental effects, it is not yet possible to define a cause-and-effect relationship between epigenomic changes and the onset and intensity of neural disorders.
Generic carvedilol 25mg with amex
Variation among members of the same inbred strain reared under different conditions is more likely to be due to environmental factors zopiclone arrhythmia buy on line carvedilol. Other approaches involve analysis of variance for a quantitative trait among offspring from different crosses, or comparing expression of a trait among offspring and parents reared in the same environment. Therefore, another type of estimate, narrow-sense heritability, has been devised that is of more practical use. Narrow-Sense Heritability Narrow-sense heritability (h2) is the proportion of phenotypic variance due to additive genotypic variance alone. Genotypic variance can be divided into subcomponents representing the different modes of action of alleles at quantitative trait loci. As not all the genes involved in a quantitative trait affect the phenotype in the same way, this partitioning distinguishes among three different kinds of gene action contributing to genotypic variance. The amount of interactive variance is often negligible, and so this component is often excluded from calculations of total genotypic variance. Few quantitative traits have very high or very low heritability estimates, suggesting that both genetics and environment play a part in the expression of most phenotypes for the trait. It does not distinguish between quantitative trait loci with alleles acting additively as opposed to those with epistatic or dominance effects. Broad-sense heritability estimates also assume that the genotype-byenvironment interaction variance component is negligible. While broad-sense heritability estimates for a trait are of general genetic interest, these limitations mean this kind of heritability is not very useful in breeding programs. Narrow-sense heritability h2 provides a more accurate prediction of selection response than broad-sense heritability H 2 and therefore h2 is more widely used by breeders. Artificial Selection Artificial selection is the process of choosing specific individuals with preferred phenotypes from an initially heterogeneous population for future breeding purposes. If selection is for a simple trait controlled by just one or two genes subject to little environmental influence, generating the desired population of plants or animals is relatively fast and easy. However, many traits of economic importance in crops and livestock, such as grain yield in plants, weight gain or milk yield in cattle, and speed or stamina in horses, are polygenic and frequently multifactorial. Narrow-sense heritability estimates are valuable to the plant or animal breeder because, as we have just seen, they estimate the proportion of total phenotypic variance for the trait that is due to additive genetic variance. Quantitative trait alleles with additive impact are those most easily manipulated by the breeder. The higher the estimated value for h2 in a population, the more likely the breeder will observe a change in phenotypic range for the trait in the next generation after artificial selection. Partitioning the genetic variance components to calculate h2 and predict response to selection is a complex task requiring careful experimental design and analysis. The simplest approach is to select individuals with superior phenotypes for the desired quantitative trait from a heterogeneous population and breed offspring from those individuals. The relationship between these means and h2 is h2 = M2 - M M1 - M are interbred, and the mean diameter M2 of the progeny kernels is 13 mm. We can calculate the realized heritability h2 to estimate the potential for artificial selection on kernel size: h2 = h2 = = M2 - M M1 - M 13 - 20 10 - 20 -7 -10 = 0. The longest running artificial selection experiment known is still being conducted at the State Agricultural Laboratory in Illinois. With each cycle of successful selection, more of the corn plants accumulate a higher percentage of additive alleles involved in oil production. Theoretically, the process will continue this equation can be further simplified by defining M2 - M as the selection response (R), which is the degree of response to mating the selected parents, and defining M1 - M as the selection differential (S), which is the difference between the mean for the whole population and the mean for the selected population. As an example of a realized heritability estimate, suppose that we measure the diameter of corn kernels in a population where the mean diameter M is 20 mm. From this population, we select a group with the smallest diameters, for which the mean M1 equals 10 mm. The numbers in parentheses at generations 9, 25, 52, and 76 for the high-oil line indicate the calculation of heritability at these points in the continuing experiment. At that point, h2 will be reduced to zero and response to artificial selection will cease. The decrease in response to selection for low oil content shows that heritability for low oil content is approaching this point. Remember, this does not indicate the absence of a genetic contribution to the observed phenotypes for such traits. Instead, the low h2 values show that natural selection has already largely optimized the genetic component of these traits during evolution. Egg production, litter size, and conception rate are examples of how such physiological limitations on selection have already been reached. Traits that are less critical to survival, such as body weight, tail length, and wing length, have higher heritabilities because more genotypic variation for such traits is still present in the population. Remember also that any single heritability estimate can only provide information about one population in a specific environment. Therefore, narrowsense heritability is a more valuable predictor of response to selection when estimates are calculated for many populations and environments and show the presence of a clear trend. Measured heritability depends on the environmental variation present in the population being studied and cannot be used to evaluate differences between populations. Twins are said to be concordant for a given trait if both express it or neither expresses it. If one expresses the trait and the other does not, the pair is said to be discordant. In some cases-for example, blood types and eye color-we know that this is indeed true. In the case of contracting measles, however, a high concordance value merely indicates that the trait is almost always induced by a factor in the environment-in this case, a virus. In the case of measles, where concordance is high in both types of twins, the environment is assumed to be the major contributing factor. Such an analysis is useful because phenotypic characteristics that remain similar in different environments are likely to have a strong genetic component. Twin Studies Have Several Limitations Interesting as they are, human twin studies contain some unavoidable sources of error. Another possible error source is interactions between the genotype and the environment that produce variability in the phenotype. Overall, heritability estimates for human traits based on twin studies should therefore be considered approximations and examined very carefully before any conclusions are drawn. These disorders clearly have genetic components, and twin studies provide a foundation for studying interactions between genes and environmental factors. Such research has also opened the way to new approaches to the study of interactions between the genotype and environmental factors. The most relevant genomic discoveries about twins include the following: Large-Scale Analysis of Twin Studies For decades, researchers have used twin studies to examine the relative contributions of genotype and environment to the phenotypic variation observed in complex traits in humans. These traits involve the interplay of multiple genes with a network of environmental factors, and the genetic components of the resulting phenotypic variance can be difficult to study. The simplest way to assess the genetic contribution is to assume that the effect of each gene on a trait is independent of the effects of other genes. Because the effects of all genes are added together, this is called the additive model. However, in recent years, some geneticists have proposed that nonadditive factors such as dominance and epistasis are more important than additive genetic effects. As a result, the relative roles of additive and nonadditive factors are a subject of active debate. In an attempt to resolve this issue, an international project recently examined the results of all twin studies performed in the last 50 years. This study, published in 2015, involved the compilation and analysis of the data for over 17,000 traits studied in more than 14 million twin pairs drawn from more than 2700 published papers. This does not exclude the role of nonadditive factors such as dominance and epistasis, but these factors most likely play a secondary role in heritability. Third, genetic variance is an important component of the individual variations observed in populations. In addition, the relative effects of genotypes and environmental factors are nonrandomly distributed, making their contributions somewhat trait-specific. In those pairs where it does occur, one estimate is that such divergence takes place in 15 to 70 percent of the somatic cells. Other complex disorders displaying a genetic component are similarly being investigated using epigenetic analysis in twin studies.
Generic carvedilol 25 mg mastercard
By contrast pulse pressure 36 cheap carvedilol 12.5mg amex, daughter cells resulting from mitosis are usually genetically identical. In angiosperms, meiosis results in the formation of microspores (male) and megaspores (female), which give rise to the haploid male and female gametophyte stage. The folded-fiber model is based on each chromatid consisting of a single fiber wound like a skein of yarn. A coiling process occurs during the transition of interphase chromatin into more condensed chromosomes during prophase of mitosis or meiosis. At the end of prophase I, maternal and paternal copies of each homologous chromosome (Am and Ap, Bm and Bp, Cm and Cp) will be synapsed. At the completion of anaphase I, eight possible combinations of products (Am or Ap, Bm or Bp, Cm or Cp) will occur. Fertilization of the gametes described in Problem 29 will give the following zygotes: Zygote 1: two copies of chromosome A two copies of chromosome B three copies of chromosome C two copies of chromosome A two copies of chromosome B one copy of chromosome C Zygote 2: None of the zygotes will be diploid. The resulting zygote would have one copy of chromosome 21 (from the father) and two copies of all the other chromosomes. For the phenotypic frequencies, set up the problem in the following manner: 3/4 B 3/4 A -1/4 bb 1/4aa-3/4 B 1/4 bb 3/4 C = 27/64 A B C 1/4 cc = 9/64 A B cc 3/4 C 1/4 cc 3/4 C 1/4 cc 3/4 C 1/4 cc etc. To deal with parts (b) and (c) it is easier to see the observed values for the monohybrid ratios if the phenotypes are listed: smooth, yellow smooth, green wrinkled, yellow wrinkled, green 315 108 101 32 4. It is naturally self- For the smooth: wrinkled monohybrid component, the smooth types total 423 (315 + 108), while the wrinkled types total 133 (101 + 32). Expected ratio 3/4 1/4 Observed (o) 423 133 Expected (e) 417 139 fertilizing, but it can be crossbred. First, two alternatives (black and white) of one characteristic (coat color) are being described; therefore, a monohybrid condition exists. Note that all the offspring are black; therefore, black can be considered dominant. The second sentence of the problem verifies that a monohybrid cross is involved because of the 3/4 black and 1/4 white distribution in the offspring. We fail to reject the null hypothesis and are confident that the observed values do not differ significantly from the expected values. When homologous chromosomes separate from each other at anaphase I, alleles will go to opposite poles of the meiotic apparatus. Different gene pairs on the same homologous pair of chromosomes (if far apart) or on nonhomologous chromosomes will separate independently from each other during meiosis. In Cross 2, a typical 3:1 Mendelian ratio is observed, which indicates that two heterozygotes were crossed: Ww * Ww 14. We would therefore say that there is a "good fit" between the observed and expected values. The only difference is that the recessive genes are coming from both parents, rather than from one parent only as in part (a). When you have genes on the autosomes (not X-linked), independent assortment, complete dominance, and no gene interaction (see later) in a cross involving double heterozygotes, the offspring ratio will be 9:3:3:1. Even though this cross involves two gene pairs, it will give a "monohybrid" type of ratio because one of the gene pairs is homozygous (body color) and one gene pair is heterozygous (wing length). As the critical p value is increased, it takes a smaller x2 value to cause rejection of the null hypothesis. It would take less difference between the expected and observed values to reject the null hypothesis; therefore, the stringency of failing to reject the null hypothesis is increased. Under that circumstance, there is a 25 percent chance that each of their children would be affected. The probability that two children of heterozygous parents would be affected would be 0. In the first cross, notice that a 3: 1 ratio exists for the spiny to smooth phenotypes, leading to the hypothesis that the spiny allele is dominant to smooth. Apply the same reasoning to the second cross, where there is a 3: 1 ratio of purple to white. The purple, spiny F1 would support the hypothesis that purple is dominant to white and spiny is dominant to smooth. In the F2, a 9: 3: 3: 1 ratio would not only support the above hypothesis, but also indicate the independent inheritance and expression of the two traits. Also, there are only three possibilities: both are heterozygous, neither is heterozygous, and at least one is heterozygous. You have already calculated the first two probabilities; the last is simply 1 - (1/12 + 6/12) = 5/12. One would reject the null hypothesis and assume a significant difference between the observed and expected values. In fact, most statisticians recommend that the expected values in each class should not be less than 10. Crosses: ckck ckck ckcd ckcd ckca ckca * * * * * * cdcd cdca cdcd cdca cdcd cdca all sepia all sepia 1/2 sepia; 1/2 cream 1/2 sepia; 1/2 cream 1/2 sepia; 1/2 cream 1/2 sepia; 1/4 cream; 1/4 albino (d) Parents: sepia * cream Because the sepia parent had a full color parent and an albino parent (Cck * caca), it must be ckca. The cream parent had two full color parents that could be Ccd or Cca; therefore, it could be cdcd or cdca. Crosses: ckca * cdcd ckca * cdca b = red 1/2 sepia; 1/2 cream 1/2 sepia; 1/4 cream; 1/4 albino 4. C chC ch = chestnut C cC c = cremello C chC c = palomino (b) the F1 resulting from matings between cremello and chestnut horses would be expected to be all palomino. The F2 would be expected to fall in a 1: 2: 1 ratio as in the third cross in part (a). F2 offspring would have the following "simplified" genotypes with the corresponding phenotypes: A C = 9/16 (agouti) A cc = 3/16 (colorless because cc is epistatic to A) aaC = 3/16 (black) aacc = 1/16 (colorless because cc is epistatic to aa) the two colorless classes are phenotypically indistinguishable; therefore, the final ratio is 9: 3: 4. Half of the pigmented offspring are black and half are agouti; therefore, the female must have been Aa. Your essay should include a description of alleles that do not function independently of each other or that reduce the viability of a class of offspring. With multiple alleles, there are more than two alternatives of a given gene at a given locus. The P allele behaves as a recessive in terms of lethality (seen only in the homozygote) but as a dominant in terms of coat color (seen in the homozygote). Because many individuals in a population could have genotypes with the i allele, one could not prove that a particular male was the father by this method. Three independently assorting characteristics are being dealt with: (1) flower color (incomplete dominance), (2) flower shape (dominant/recessive), and (3) plant height (dominant/recessive). Phenotypes: gray * gray (c) Notice that 16/64 or 1/4 of the offspring are albino; therefore, the parents are both heterozygous at the C locus. Second, notice that without considering the C locus, there is a 27: 9: 9: 3 ratio that reduces to a 9: 3: 3: 1 ratio. F2: Phenotypes: 3/16 normal females 3/16 normal males 1/16 ebony females 1/16 ebony males 3/16 scalloped females 3/16 scalloped males 1/16 scalloped, ebony females 1/16 scalloped, ebony males Forked-line method: P1: F1: F2: Wings 1/4 1/4 1/4 1/4 females, normal females, scalloped males, normal males, scalloped Color 3/4 normal 1/4 ebony 3/4 normal 1/4 ebony 3/4 normal 1/4 ebony 3/4 normal 1/4 ebony 3/16 1/16 3/16 1/16 3/16 1/16 3/16 1/16 X sd X sd; e +/e + * X +/Y; e/e 1/2 X +X sd; e +/e (female, normal) 1/2 X sd/Y; e +/e (male, scalloped) 20. Notice that the X symbol may remain to remind us that the sd gene is on the X chromosome. It is extremely important that one account for both the mutant genes and each of their wild-type alleles. Phenotypic expression is dependent on the genome of the organism; the immediate molecular and cellular environment of the genome; and numerous interactions between a genome, the organism, and the environment. The dominant genes are on one homolog, while the recessive alleles are on the other homolog. Given the degree of outcrossing, that the gene is probably quite rare and therefore heterozygotes are uncommon, and that the frequency of transmission is high, it is likely that this form of male precocious puberty is caused by an autosomal dominant, sex-limited gene. Your essay should include methods of detection through crosses with appropriate, distinguishable markers and that, in most cases, the frequency of crossing over is directly related to the distance between genes. With some qualification, especially around the centromeres and telomeres, one can say that crossing over is somewhat randomly distributed over the length of the chromosome. Two loci that are far apart are more likely to have a crossover between them than two loci that are close together. If the probability of one event is 1/X then the probability of two events occurring at the same time will be 1/X 2 8. Each cross must be set up in such a way as to reveal crossovers (c,d) the coefficient of coincidence = 0. It is necessary that genetic heterogeneity exists so that different arrangements of genes, generated by crossing over, can be distinguished.
Generic carvedilol 6.25mg
In the next section we discuss the central role of bioinformatics in this process arrhythmia ekg strips buy 6.25mg carvedilol with visa. Databases are essential for archiving and sharing data with other researchers and with the public. GenBank shares and acquires data from databases in Japan and Europe; it contains more than 220 billion bases of sequence data from over 100,000 species; and it doubles in size roughly every 18 months! As sequences are identified and genes are named, each sequence deposited into GenBank is provided with an accession number that scientists can use to access and retrieve that sequence for analysis. Genome projects accumulate nucleotide sequences, and then scientists have to make sense of those sequences. Thus, after a genome has been sequenced and compiled, scientists are faced with the task of identifying gene-regulatory sequences and other sequences of interest in the genome so that gene maps can be developed. This process, called annotation, relies heavily on bioinformatics, and a wealth of different software tools are available to carry it out. The rat contig sequence was used as a query sequence to search a mouse database in GenBank. Notice that the two sequences show 93 percent identity, strong evidence that this rat contig sequence contains a gene for the insulin receptor. Gaps, indicating missing bases in the two sequences, are usually ignored in calculating similarity scores. The aligned rat and mouse sequences were 93 percent identical and showed no gaps in the alignment. Shorter sequences have a much greater likelihood of being present in the database by chance than longer sequences. The lower the E-value (the closer it is to 0), the higher the significance of the match. Because this mouse sequence on chromosome 8 is known to contain an insulin receptor gene (encoding a protein that binds the hormone insulin), it is highly likely that the rat contig sequence also contains an insulin receptor gene. We discussed many of these characteristics of a "typical" gene earlier in the text (see Chapters 13 and 17). For instance, gene-regulatory sequences found upstream of genes are marked by identifiable sequences such as promoters, enhancers, and silencers. Annotation can sometimes be a little bit easier for bacterial genes than for eukaryotic genes because there are no introns in bacterial genes. These programs incorporate search elements for many of the characteristics noted in figure 21. In fact, a reasonable question whenever one sequences a genome is, "Where are the genes Most eukaryotic genes are organized into coding segments (exons) and noncoding segments (introns). By convention, the sequence is presented in groups of ten nucleotides, although in reality the sequence is continuous. From a casual inspection, it is not clear whether this sequence contains any genes and, if so, how many. Genetic information is encoded in groups of three nucleotides (triplets), but it is not always clear whether to begin the analysis of a sequence at the first nucleotide, the second, or the third. Using this sequence as the query in a search of genomic databases would reveal that it is part of a single gene, the human b@globin gene. Thus annotation can be used to predict the number of proteins encoded by a genome. Prediction programs can also search for codon bias, the more frequent use of one or two codons to encode an amino acid that can be specified by a number of different codons. If the codons were used randomly, each would be used about 25 percent of the time. Codon bias is present in exons but should not be present in introns or intergenic spacers. Functional genomics can involve experimental approaches to confirm or refute computational predictions (such as the number of protein-coding genes). Inferring gene function from similarity searches is based on a relatively simple idea. If a genome sequence shows statistically significant similarity to the sequence of a gene whose function is known, then it is likely that the genome sequence encodes a protein with a similar or related function. Another major benefit of similarity searches is that they are often able to identify homologous genes, genes that are evolutionarily related. In the globin gene family, the a@ and b@globin subunits in humans are paralogs resulting from a geneduplication event. If homologous genes in different species are thought to have descended from a gene in a common ancestor, the genes are known as orthologs. For instance, mouse and human a@globin genes are orthologs evolved from a common ancestor. As an interesting aside, the leptin gene (also called Lep, for leptin, in mice) is highly expressed in fat cells (adipocytes). This gene produces the protein hormone leptin, which targets cells in the brain to suppress appetite. Although it is important to note that weight control is not regulated by a single gene, the discovery of leptin has provided significant insight into lipid metabolism and weight disorders in humans. Predicting Function from Structural Analysis of Protein Domains and Motifs When a gene sequence is used to predict a polypeptide sequence, the polypeptide sequence can be analyzed for specific structural domains and motifs. These motifs can often easily be searched for using bioinformatics software, and their identification in a sequence is a common strategy for inferring the possible functions of a protein. Investigators Are Using Genomics Techniques Such as Chromatin Immunoprecipitation to Investigate Aspects of Genome Function and Regulation In this chapter and later in the text (see Chapter 22), we will consider a range of different genomic techniques that are valuable for functional genomics studies. This allows researchers to study an entire genome to locate binding sites for proteins such as transcription factors, histone-related proteins, and other proteins involved in chromatin structure. Notice from the number of identical nucleotides, indicated by shaded boxes and vertical lines, that the nucleotide sequence for these two genes is very similar. It has produced a plethora of information, much of which is still being analyzed and interpreted. What is already clear, based on all the different kinds of genomes that have been sequenced, is that humans and all other species share a common set of genes essential for cellular function and reproduction, confirming that all living organisms arose from a common ancestor. It established a 15-year plan with a proposed budget of $3 billion to identify all human genes, originally thought to number between 80,000 and 100,000, to sequence and map them all, and to sequence the approximately 3 billion base pairs thought to be comprised by the 24 chromosomes (22 autosomes, plus X and Y) in humans. Map and sequence the genomes of several model organisms used in experimental genetics, including Escherichia coli, Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster, and Mus musculus (mouse). Develop new sequencing technologies, such as highthroughput computer-automated sequencers, to facilitate genome analysis. Much of the work was carried out by the International Human Genome Sequence Consortium, involving nearly 3000 scientists working at 20 centers in six countries (China, France, Germany, Great Britain, Japan, and the United States). Craig Venter at Celera Genomics (aptly named from a word meaning "swiftness") was announced. The public project had proposed using a clone-by-clone approach to sequence the genome. The assembled sequence consists of haploid genomes pooled from different individuals so that they provide a reference genome representative of major, common elements of a human genome widely shared among populations of humans. As you can see in this table, many unexpected observations have provided us with major new insights. Genome variations, including the abundance of repetitive sequences scattered throughout the genome, verify that the genome is dynamic, revealing many evolutionary examples of sequences that have changed in structure and location. The average size of a human gene is 25 kb, including generegulatory regions, introns, and exons. Many human genes produce more than one protein through alternative splicing, thus enabling human cells to produce a much larger number of proteins (perhaps as many as 200,000) from only 20,000 genes. More than 50 percent of human genes show a high degree of sequence similarity to genes in other organisms; however, more than 40 percent of the genes identified have no known molecular function. Gene-rich clusters are separated by gene-poor "deserts" that account for 20 percent of the genome. Chromosome 19 has the highest gene density, and chromosome 13 and the Y chromosome have the lowest gene densities. Chromosome 1 contains the largest number of genes, and the Y chromosome contains the smallest number. Human genes are larger and contain more and larger introns than genes in the genomes of invertebrates, such as Drosophila. The number of introns in human genes ranges from 0 (in histone genes) to 234 (in the gene for titin, which encodes a muscle protein).
